La Patología y la Biología
Figura 1
El SS es clínica, morfológica y genéticamente distinto al resto de los sarcomas. Se caracteriza por una translocación cromosómica específica (X, 18) (p11, q11). En la última 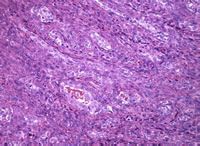 clasificación de la OMS sobre tumores óseos y de partes blandas, el SS está clasificado entre los tumores malignos de diferenciación incierta, sin correspondencia con algún tejido normal (OMS 2002). Incluso siendo típico de tejidos blandos como el músculo, el SS puede encontrarse también en vísceras, como el riñón, el pulmón y la pleura.
clasificación de la OMS sobre tumores óseos y de partes blandas, el SS está clasificado entre los tumores malignos de diferenciación incierta, sin correspondencia con algún tejido normal (OMS 2002). Incluso siendo típico de tejidos blandos como el músculo, el SS puede encontrarse también en vísceras, como el riñón, el pulmón y la pleura.
Los SS se clasifican en función de su aspecto morfológico como:
SS bifásico
SS monofásicos fusiformes
SS monofásicos epitelioides (excepcionales)
SS pobremente diferenciados
Translocaciones: Significa la rotura de una porción de cromosoma, que se inserta en otro cromosoma diferente. El SS tiene una translocación cromosómica característica entre el cromosoma X y el 18, que se considera fundamental para su desarrollo. La existencia de una translocación determinada proporciona una diana potencial para el desarrollo de tratamientos dirigidos.
Los SS bifásicos muestran células tanto fusiformes como epitelioides (Figura 1).
Los SS monofásicos presentan sólo el componente de células fusiformes (Figura 2). El SS 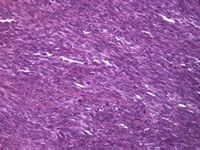 monofásico epiteloide es identico en su aspecto a los adenocarcinomas (distintos de los sarcomas), de los que solo puede distinguirse mediante el empleo de técnicas diagnósticas muy especiales de genética molecular.
monofásico epiteloide es identico en su aspecto a los adenocarcinomas (distintos de los sarcomas), de los que solo puede distinguirse mediante el empleo de técnicas diagnósticas muy especiales de genética molecular.
Los SS pobremente diferenciados muestran uno de tres patrones morfológicos: células grandes, células pequeñas, o un patrón especial de células fusiformes mal definidas
Entre los sistemas de clasificación disponibles, el sistema francés, FNCLCC (Federación Francesa de Centros de Cáncer del Grupo de Sarcomas), es el más empleado para los sarcomas de partes blandas de los adultos (Guillou, 1997). Se trata de un sistema de  puntuación en el que la suma de los valores asignados a D (diferenciación, ‘D3’ por defecto en el SS), M (mitosis: por debajo de 10 mitosis / 10 campos de gran aumento; entre 10 y 19/10 CGA, más de 19/CGA) y N (necrosis: ausente, menos de 50%, más del 50%) da el grado 1, 2 o 3. Incluso si en los protocolos terapéuticos SS es considerado como uno de sarcomas de alto grado, por definición, el grado de FNCLCC parece ser el factor histológico más predictivo de metástasis en los SS. Las características que confieren un mejor pronóstico a los SS son tener menos de 10 mitosis/10CGA, la ausencia de necrosis, la ausencia de áreas pobremente diferenciadas, la edad pediátrica, el tamaño menor de 5 cm y la extirpación completa de un tumor localizado (Guillou, 2004).
puntuación en el que la suma de los valores asignados a D (diferenciación, ‘D3’ por defecto en el SS), M (mitosis: por debajo de 10 mitosis / 10 campos de gran aumento; entre 10 y 19/10 CGA, más de 19/CGA) y N (necrosis: ausente, menos de 50%, más del 50%) da el grado 1, 2 o 3. Incluso si en los protocolos terapéuticos SS es considerado como uno de sarcomas de alto grado, por definición, el grado de FNCLCC parece ser el factor histológico más predictivo de metástasis en los SS. Las características que confieren un mejor pronóstico a los SS son tener menos de 10 mitosis/10CGA, la ausencia de necrosis, la ausencia de áreas pobremente diferenciadas, la edad pediátrica, el tamaño menor de 5 cm y la extirpación completa de un tumor localizado (Guillou, 2004).
Aspecto macroscópico: El diámetro de los SS suele variar entre 3 y 10 cm. Los tumores tienden a ser multinodulares y pueden ser quísticos. Cuando crecen lentamente, suelen comprimir el tejido circundante, formando una pseudo-cápsula. Los SS pobremente diferenciados son más agresivos, crecen rápidamente, carecen de pseudo-cápsula, sus márgenes son infiltrantes, y pueden mostrar hemorragia y necrosis.
Aspecto microscópico: Los SS se componen de dos tipos de células de aspecto y características distintas: las células fusiformes (con forma de huso), uniforme y relativamente pequeñas, con núcleos ovales y escaso citoplasma, formando láminas sólidas, y las células epitelioides (redondas o poligonales).
Sarcoma Sinovial Pobremente Diferenciado se considera como una forma de progresión, con un comportamiento más agresivo y una mayor probabilidad de originar metástasis (Weiss, 2001). En las zonas menos celulares puede haber hialinización, cambios rizoides ni calcificaciones, con o sin la osificación y, rara vez, cambios condroides. La calcificación tumoral focal, con o sin osificación, está presente en cerca de un tercio de los SS pobremente diferenciados.
Genética molecular: Los SS se caracterizan la presencia de una translocación específica t (X, 18), que fusiona el gen SYT del cromosoma 18, con el gen SSX1 (aproximadamente 2 / 3 de los casos), SSX2 (aproximadamente 1 / 3 de los casos ) o SSX4 (casos raros) del cromosoma X. Como consecuencia de la translocación, la célula maligna sintetiza una proteína nueva detectable por una técnica especial conocida como PCR. Se han descrito casos tanto con fusiones SYT/SSX1 como SYT/SSX2. Algunos informes señalan que SYT/SSX1 se asocia con el SS bifásico. Puede que la fusión SYT/SSX1 se relacione con una mayor incidencia de metástasis, pero esto no ha sido completamente confirmado, por lo que la importancia pronóstica de la tipificación del gen de fusión es todavía incierta (Mancuso 2000, Mezzelani 2001).
Características Clínicas y Diagnóstico de los Sarcomas Sinoviales
Los SS pueden surgir en cualquier lugar del cuerpo, presentándose generalmente como una masa en progresivo aumento. La presentación clínica más frecuente es una masa de lento crecimiento en los tejidos blandos de las extremidades inferiores, especialmente alrededor de la rodilla y el tobillo. El tumor está a menudo cerca de un tendón. Son menos comunes en otros lugares como la cabeza, el cuello, la pared abdominal, el retroperitoneo o fondo de la espalda, el mediastino o centro del pecho, la pleura o los pulmones.
Los síntomas pueden ser muy variados, aunque la masa indolora sigue siendo la presentación más frecuente. La dificultad para la deglución y la respiración, o la alteración de la voz, por ejemplo, podrían estar asociados con el SS de la región de cabeza y cuello. El dolor puede estar relacionado con la afectación de los nervios. Debido a que el tumor crece lentamente, los síntomas pueden estar presentes durante largo tiempo antes de que se haga el diagnóstico, lo que puede retrasarlo.
Como se trata de un sarcoma de alto grado, los SS se caracteriza por la invasión local y una propensión a originar metástasis o ramificaciones a distancia. En el momento del diagnóstico, menos del 10% de los casos se presentan con metástasis (especialmente en los pulmones), pero la diseminación posterior, se puede producir en el 25-50% de los casos.
Con el fin de determinar la extensión de la enfermedad, es necesario un estudio completo de imagen. Estos estudios son fundamentales para determinar el tamaño del tumor y la extensión local. La 3cografía es, a menudo, la primera herramienta que se 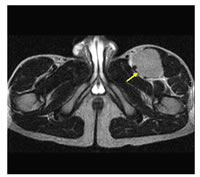 utilizar. La tomografía computarizada (TAC) o La Resonancia Magnética (RM) de la localización primaria es obligatoria para la evaluación de la extensión local antes de cualquier tratamiento. La RM de una extremidad se suele considerar la mejor prueba para definir la extensión de los tumores en los tejidos blandos (Figura 4).
utilizar. La tomografía computarizada (TAC) o La Resonancia Magnética (RM) de la localización primaria es obligatoria para la evaluación de la extensión local antes de cualquier tratamiento. La RM de una extremidad se suele considerar la mejor prueba para definir la extensión de los tumores en los tejidos blandos (Figura 4).
Después de la descripción precisa de la extensión local del tumor, la evaluación patológica es necesaria para definir el diagnóstico histológico. La biopsia inicial tiene el objetivo de definir el diagnóstico, pero también debe proporcionar suficiente material para la inmunohistoquímica, la citogenética, los estudios biológicos y la revisión centralizada de la patología en aquellos pacientes incluidos en ensayos clínicos multicéntricos.
En el caso de una masa grande y profunda, la biopsia debe ser siempre el procedimiento quirúrgico inicial, a fin de evitar la cirugía inadecuada. La biopsia abierta (biopsia incisional) o biopsia con aguja gruesa (tru-cut, guiada por ultrasonido o por tomografía computarizada) es preferible a los aspirados con aguja fina, capaces de establecer la presencia de malignidad, pero rara vez de identificar el subtipo o proporcionar el tejido necesario para estudios adicionales. En cualquier caso, la biopsia inicial debe ser cuidadosamente planificada por un cirujano experimentado, teniendo en cuenta la posible cirugía definitiva posterior, que debe incluir la cicatriz y el trayecto de la biopsia. Por ejemplo, en SS de las extremidades, la incisión debe ser longitudinal a la extremidad y no atravesar varios compartimentos; Se debe de tener mucho cuidado y se debe minimizar el riesgo de hematomas post-quirúrgicos y la necesidad de drenajes. En todos los casos, el tejido debe ser enviado en estado fresco al laboratorio. El fijador que se utilizará cuando esto no sea posible, será el formol.
Estadificación
Después de la evaluación radiológica de la extensión del tumor y de la biopsia, el estudio diagnóstico se completa con otras pruebas cuyo fin es el de detectar metástasis, si las hubiera. El pulmón, el esqueleto y los órganos abdominales se registran mediante TAC, gammagrafía ósea con tecnecio y ecografía abdominal . Es también importante la atención a los ganglios linfáticos próximos al sarcoma. La tomografía por emisión de positrones (PET) no se considera una prueba de imagen útil para el SS.
La estrategia de tratamiento se basa en la estadificación obtenida en base a todas estas pruebas, tanto antes como después de la cirugía. Los oncólogos pediátricos suelen estadificar a los SS, según la clasificación TNM (La T señala el tamaño del tumor por encima o debajo de los 5 cm, la N la afectación de los ganglios, y la M la existencia de metástasis) (Harmer 1982).
La clasificación del Intergrupo de Estudio del Rabdomiosarcoma (IRS) se basa en el grado de extirpación quirúrgica (Maurer, 1988):
Grupo I – Extirpación completa con márgenes negativos microscópicos
Grupo II – Extirpación amplia, pero con residuos microscópios y/o afectación de los ganglios
Grupo III – Extirpación incompleta, con restos visibles de la enfermedad.
Grupo IV – Metástasis de inicio.
Los oncólogos de adultos suelen usar el sistema de clasificación del Comité Conjunto Americano del Cáncer, que incorpora el grado histológico (que es alto por definición en todos los pacientes con SS), además del tamaño y la profundidad (la mayoría de los SS son profundos).
Pronóstico
El pronóstico de los pacientes con SS se relaciona con la posibilidad de extirparlo por completo, el tamaño del tumor, y la invasión local. Los pacientes con tumores pequeños que se pueden eliminar completamente en el momento del diagnóstico tienen un pronóstico excelente. Para los tumores mayores de 5 cm, el riesgo de desarrollar metástasis a distancia es mayor. La serie Pediátrica (Ladenstein de 1993, Pappo 1994, Ferrari 1999, Okcu 2003, Brecht, en prensa) observó una supervivencia del 80% para los pacientes del grupo I-II de la IRS, pero alrededor del 60-70% de los casos> 5 cm. La supervivencia de los pacientes con tumores inextirpables al momento del diagnóstico (IRS grupo III), alcanzan una supervivencia entre el 50 y el 70%, claramente menor para los casos de la región del cuello-cabeza, pulmón y abdomen. Los pacientes con metástasis a distancia tienen un pronóstico muy pobre.
El Tratamiento del Sarcoma Sinovial
El enfoque del tratamiento óptimo del SS no está completamente claro. Como en otros sarcomas de partes blandas, el tratamiento estándar para la enfermedad localizada es la cirugía. La radioterapia tiene un papel en la mejora del control local después de la resección compartimental. El papel de la quimioterapia aún no está claro y la rareza de estos tumores dificulta reunir de un número adecuado de pacientes para llevar a cabo ensayos clínicos de calidad. Sin embargo, es posible afirmar que la resección quirúrgica con o sin la radioterapia adyuvante y/o quimioterapia basada en doxorrubicina e ifosfamida son los pilares actuales de tratamiento.
Cirugía
La cirugía es la piedra angular del tratamiento del SS. Su objetivo es obtener márgenes adecuados con pocas o ninguna secuela a largo plazo, y, en la medida de lo posible, la amputación. Si este objetivo no parece alcanzable en un primer momento, es posible administrar quimioterapia y/o radioterapia para reducir el tumor y poderlo someter a cirugía posteriormente. El logro de márgenes adecuados es una cuestión crucial, estrictamente influenciada por el tipo de tejido sano alrededor del tumor. Es muy difícil cuantificar con exactitud la distancia que se considera “segura” entre el tumor y los márgenes de resección (Gronchi 2005). La conclusión de la Conferencia de Sarcomas de Partes Blandas en Adultos, celebrada en Milán en junio de 2004 sugirió como márgenes adecuados lo que son “mayores de 1 cm de tejido sano alrededor del tumor, en todas las direcciones, cuando el tejido es un músculo, y de 1 mm de tejido sano alrededor el tumor cuando el tejido una barrera anatómica, como el periostio, (tejido mesenquimatoso que envuelve los huesos, excepto en las articulaciones), los vasos, el epineuro, o la fascia muscular.
Los márgenes quirúrgicos inadecuados afectan negativamente a los resultados locales y, por lo tanto, también a la supervivencia (aunque algunos estudios sarcomas de partes blandas de adultos no encontraron una fuerte correlación entre la calidad de la cirugía y el resultado final respecto a la supervivencia). Sin embargo, la cirugía adecuada se podría definir una resección R0 que hace que los pacientes sean clasificados como Grupo I. Esto incluye tanto las resecciones compartimentales (extirpación en bloque del tumor y de todo el compartimiento muscular de origen, si el tumor estaba totalmente confinado en él) como las resecciones amplias (extirpaciones en bloque a través del tejido normal, incluyendo todo el tumor con su pseudocápsula).
Cirugía: Cuándo y Dónde – La calidad de la intervención quirúrgica es fundamental. Las masas de partes blandas grandes (es decir, de más de 5 cm) y profundas tienen una alta probabilidad de tratarse de un sarcoma y deben ser remitidos a centros especializados para la cirugía, idealmente, antes incluso de realizar la biopsia.
Quimioterapia
En cuanto al papel de la quimioterapia, es bastante sorprendente que, a lo largo de los años, se han establecido estrategias diferentes en los niños y en los adultos. Dado que es en los niños en los que se han observado tasas muy altas de respuesta a la quimioterapia, los oncólogos pediátricos han equiparado el SS al rabdomiosarcoma, considerándolo un tumor quimiosensible. Por lo tanto, los pacientes pediátricos han recibido quimioterapia adyuvante, independientemente del estadio del sarcoma, incluso después de la extirpación completa de tumores muy pequeños (Ladenstein de 1993, Pappo de 1994, y Ferrari, 1999). En cambio, en el caso de los adultos, la quimioterapia adyuvante sólo se ha utilizado en ensayos clínicos que incluían, además del SS, muchas otras variedades de sarcoma (Meta Análisis Cooperativo de Sarcomas 2997, Bergh 1999, Lewis 2000, Spillane 2000, Frustaci de 2001, y Trassard 2001 ). Sólo recientemente, los oncólogos de adultos empiezan a considerar el posible papel de la quimioterapia adyuvante para los casos de alto riesgo, es decir, cuando el tamaño del tumor es grande (Frustaci 2001, Spurrel 2005).
Un análisis multivariante retrospectivo multicéntrico coordinado por el MD Anderson Cancer Center, que incluye los resultados actualizados de la serie pediátrica, examinó la historia clínica y la estrategia de tratamiento de los niños y adolescentes con SS (Okcu 2003). La supervivencia global (OS) de los 219 pacientes fue del 80% a los 5 años, superior a la registrada en la serie de adultos, y la tasa de respuesta a la quimioterapia alcanzó el 60%, mayor que la obtenida normalmente en los sarcomas de adultos. Sin embargo, el análisis sugiere que la quimioterapia adyuvante no tuvo impacto sobre la supervivencia. La supervivencia libre de eventos (SLE) fue del 84% para los 37 pacientes sin tratamiento y el 78% entre los 122 pacientes tratados con quimioterapia adyuvante (Okcu 2003).
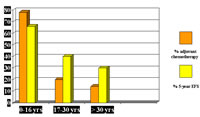 Figura 5
Figura 5
Otro estudio del Instituto Nacional de Tumores de Milán, con 271 pacientes, comparó los resultados clínicos, las modalidades de tratamiento y los resultados de los SS en las diferentes edades (Ferrari, 2004). No se observaron diferencias importantes en la presentación clínica, a excepción de una tendencia a un mayor tamaño de los tumores en los pacientes de mayor edad. Esto sugiere que no existen diferencias biológicas importantes según la edad. Sin embargo, existía una fuerte correlación de las tasas de supervivencia con los grupos de edad y el uso de la quimioterapia. La supervivencia libre de metástasis (MFS) fue del 60% en los pacientes que recibieron quimioterapia y el 48% en aquellos que no recibieron el tratamiento. Asimismo, la MFS pasó del 69% al 53% y al 43% entre las cohortes de 0-16 años (78% recibieron quimioterapia), 17-30 años (21% quimioterapia) y > 30 años (15%, quimioterapia) , véase la Figura 5 (Ferrari, 2004). Por supuesto, este análisis retrospectivo no puede interpretarse como una demostración formal de la eficacia de la quimioterapia adyuvante en las SS, pero no obstante, sugiere que tiene un papel.
Respuesta a la Quimioterapia
La respuesta a la quimioterapia del sarcoma sinovial parece equiparable a la de la mayoría de los sarcomas de partes blandas típicos de los adultos; mientras que, en los niños, se asemeja más a la de los tumores de células pequeñas y redondas, como el rabdomiosarcoma o sarcoma de Ewing. La tasa de respuesta a la quimioterapia es, en efecto, aproximadamente el 60% para el sarcoma sinovial, menos del 40% para los sarcomas de partes blandas de adultos y del 80% para el rabdomiosarcoma.
Aunque puede ser cierto que los resultados clínicos desfavorables afecten más a los adultos que a los niños y que la edad, per se, constituya un factor pronóstico para los sarcomas de partes blandas, es poco probable que la biología del SS sea esencialmente diferente en los adultos respecto a los niños. Por lo tanto, no hay razón para tratar la misma enfermedad en la misma fase de maneras diferentes según la edad del paciente.
Un análisis más detallado proviene de los Grupos Cooperativos pediátricos italiano y alemán, en los que se revisaron retrospectivamente los datos de 150 pacientes con SS resecado (Bertolt Brecht, en prensa). Los datos de este estudio no aportaron datos relevantes sobre el papel de la quimioterapia adyuvante, dado que muy pocos la recibieron. Sin embargo, identificó un subgrupo de pacientes con riesgo muy bajo de desarrollar metástasis: solo cuatro recidivas locales no metastásicas se observaron con tumores <5 cm.
Figura 6
Los datos anteriores fueron tenidos en cuenta en el desarrollo del protocolo del recientemente creado Grupo Europeo de Estudio del Sarcoma Pediátrico. El protocolo  europeo, en primer lugar, está específicamente diseñado para sarcomas de tejidos blandos distintos del rabdomiosarcoma. Este protocolo va a reclutar pacientes en toda Europa. Se fundamente en las experiencias anteriores con SS pediátricos (que lo asemejaban al rabdomiosarcoma y, por lo tanto, empleaban la quimioterapia adyuvante) y las experiencias en adultos (donde la quimioterapia adyuvante con ifosfamida y doxorubicina se usó poco). La quimioterapia se omitirá en el grupo I del IRS (pacientes con tumores menores de 5 cm), mientras que para los otros subgrupos se emplea una quimioterapia intensiva compuesta por doxorrubicina e ifosfamida, véase la figura 6 (Ferrari, 2005).
europeo, en primer lugar, está específicamente diseñado para sarcomas de tejidos blandos distintos del rabdomiosarcoma. Este protocolo va a reclutar pacientes en toda Europa. Se fundamente en las experiencias anteriores con SS pediátricos (que lo asemejaban al rabdomiosarcoma y, por lo tanto, empleaban la quimioterapia adyuvante) y las experiencias en adultos (donde la quimioterapia adyuvante con ifosfamida y doxorubicina se usó poco). La quimioterapia se omitirá en el grupo I del IRS (pacientes con tumores menores de 5 cm), mientras que para los otros subgrupos se emplea una quimioterapia intensiva compuesta por doxorrubicina e ifosfamida, véase la figura 6 (Ferrari, 2005).
Radioterapia
La radioterapia juega un papel bien definido en el control local de los sarcomas de partes blandas. En los adultos, se recomienda la irradiación después de la extirpación incompleta incompleta, pero, a menudo, también después de la excisión amplia, especialmente en el caso de tumores de gran tamaño. Ciertamente la indicación de la radioterapia tiene que ser más estricta en los niños, jóvenes y adolescentes que en los adultos, dado el mayor riesgo de secuelas sobre el crecimiento.
La serie anteriormente citada del INT de Milán informó sólo de una tendencia favorable para la adición de la radioterapia en pacientes que inicialmente fueron sometidos a resección completa. : en los pacientes del grupo I del IRS, la supervivencia libre de recidiva local (LRFS) a los 5 años fue del 78% en el grupo tratado, frente al 67% de los que no recibieron radioterapia postoperatoria. Sin embargo, sí se observó un beneficio claro en los pacientes cuya resección inicial fue marginal: el grupo II del IRS radiado obtuvo un LRFS del 57% a los 5 años, frente a solo el 7% cuando no se aplicaba la radioterapia (Ferrari, 2004). Según el protocolo EpSSG, la cirugía se debe considerar como el único tratamiento local para los pacientes del grupo I (a pesar de que la necesidad de la irradiación en el caso de los tumores mayores de 5 cm es todavía una cuestión abierta), mientras que la radioterapia post-operatoria es necesario para los pacientes del grupo II del IRS (Ferrari, 2005).
La estrategia del tratamiento local es más complicada en los pacientes cuyos tumores son irresecables al diagnóstico, y que deben recibir quimioterapia en primer lugar. Para ellos, el tratamiento de elección es demorar la cirugía y realizar todos los esfuerzos necesarios para obtener la resección completa. Sin embargo, la necesidad de radioterapia adicional tras el retraso de la cirugía de resección completa sigue siendo una cuestión sin resolver. También hay dudas sobre el momento óptimo de la radioterapia: la postoperatoria conlleva un menor riesgo de complicaciones, pero la pre-operatoria puede mejorar las posibilidades de obtener unos márgenes de resección adecuados, reducir el riesgo de contaminación intraoperatoria, y permite emplear dosis más bajas y campos más reducidos. Creemos que la elección del tratamiento local debe ser discutido en un marco multidisciplinario y la decisión ha de ser personalizado teniendo en cuenta factores deben como el sitio anatómico, el tamaño del tumor y la edad del paciente.
El Futuro
En cuanto a otros sarcomas de partes blandas, esperamos mejorar nuestra comprensión del SS en los próximos años. En particular, se necesitan enfoques terapéuticos novedosos. La translocación cromosómica específica del SS (Kawaguchi, 2005), así como los receptores de tirosin-quinasa implicados (EGFR y HER-2/neu) (Tamborini 2004, Thomas 2005), pueden ser objeto de nuevos agentes moleculares específicamente diseñados para influir en la biología del tumor (Albritton 2005). Ya se han puesto en marcha algunos ensayos clínicos con terapias dirigidas. Son también necesarios más estudios para investigar el papel de la terapia con oligonucleótidos antisentido de la Bcl-2, ya que la mayoría de los SS sobreexpresa la proteína anti-apoptótica Bcl-2, relacionada con el crecimiento del tumor y la resistencia a la quimioterapia (Mancuso 2000).
Terapias dirigidas: El sarcoma sinovial es uno de los sarcomas en los que más real y próxima parecen las terapias dirigidas contra la proteína de fusión producida por la translocación específica o contra los receptores de tirosín-quinasa sobreexpresados por las células tumorales.
El desarrollo de ensayos cooperativos que incluyan tanto pacientes pediátricos como adultos, podría ser la estrategia adecuada para acelerar los estudios biológicos y obtener, en una enfermedad rara como el SS, la cantidad de casos necesarios para diseñar ensayos clínicos adecuados.
