Con el término de sarcoma de Ewing se incluye un grupo o familia de tumores (EFT, Ewing’s family of tumors, por sus siglas en inglés) que engloba el tumor óseo de Ewing, el tumor extraóseo de Ewing, el tumor de Ewing atípico, los neuroepiteliomas periféricos o tumores neuroectodérmicos primitivos periféricos (pPNET), el tumor de Askin, que comprende un grupo de neoplasias de localización toracopulmonar, aunque también se han descrito casos con diferenciación rabdosarcomatosa embrionaria. Este grupo te tumores están muy relacionados entre si y generalmente son difíciles de distinguir .
Además de compartir algunos hallazgos clínicos y morfológicos, estas neoplasias tienen características genéticas e inmunofenotípicas que avalan su consideración dentro de una única categoría. Los avances en la comprensión de la biología básica de esta familia de tumores tienen un impacto directo en su diagnóstico y tratamiento.
El sarcoma de Ewing/pPNET es el prototipo de tumor maligno de células redondas, pequeñas y azules que afectan preferentemente al hueso, aunque también pueden afectar a partes blandas y otros órganos.
En el año 1921 James Ewing describió el endotelioma difuso del hueso, y descubrió que esta tumoración era radiosensible. En el año 1924 se le dio el nombre de mieloma endotelial. En el año 1967, debido a la elevada mortalidad, Boyer sugiere evitar la cirugía; sin embargo el progreso actual permite que el complemento de cirugía, radioterapia y quimioterapia tengan resultados superiores a 30 años atrás. En el año 1969 Johnson y Humphreys introducen la ciclofosfamida y en 1973 Rosen utiliza múltiples agentes (doxirrubicina, vincristina, ciclofosfamida y actinomicina D), logrando un esbozo de la actual quimioterapia asociada.
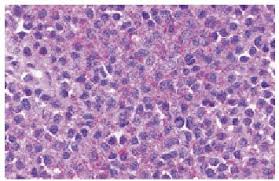
La organización mundial de la Salud (OMS) define el sarcoma de Ewing como un tumor maligno con aspecto histológico más bien uniforme, caracterizado por células pequeñas muy abundantes de núcleo redondo sin nucléolo prominente, que puede dividirse en lóbulos mediante tabiques fibrosos, sin red reticular de fibras de reticulina (a diferencia del linfoma), con o sin zonas de hemorragia y/o necrosis en su interior, y con mitosis infrecuentes.
La etiología del sarcoma de Ewing es aún desconocida. Existe traslocación parcial entre los cromosomas 11 y 22, característica compartida con algunos tumores neurales como el neuroepitelioma periférico y el neuroblastoma, lo que apoya a quienes postulan un origen neural. Otros piensan que se origina en células indiferenciadas pluripotenciales similares a osteoblastos inmaduros.
Epidemiología
Es conocido que los tumores óseos malignos son de baja frecuencia, y aunque el sarcoma de Ewing ocupa el segundo lugar, sólo representa entre el 6 y el 11% de éstos. Su incidencia anual es de 0,7 por cada millón de habitantes, es el segundo tumor óseo maligno primario más frecuente en niños luego del osteosarcoma, afecta más a hombres en una relación 1,6:1, y se presenta en la segunda década de la vida en un 65% de los casos, con un margen que va desde los 0 a los 83 años, siendo raro en menores de 5 y mayores de 30. Es muy poco frecuente en la raza negra.
En un texto clásico, en una casuística de 272 sarcomas de Ewing, el 90% de los casos ocurrió en las dos primeras décadas, especialmente durante la segunda, disminuyendo drásticamente a edades mayores; sin embargo, se observaron casos incluso en la sexta década de la vida. El grupo europeo cooperativo para el estudio del sarcoma de Ewing (European Group Cooperative Ewing’s Sarcoma Study), durante los años 1980 y 1997, con una casuística de 945 casos, precisa que el 60% de ellos ocurre en pacientes de sexo masculino y que el 57% ocurre en la segunda década, con una incidencia anual de tres casos por cada millón de habitantes menores de 15 años. En el 32% de los casos se afectan los huesos largos de las extremidades inferiores y la mitad de éstos ocurre en el fémur.
El sarcoma de Ewing se localiza en la diáfisis o en la unión diáfisometafisiaria de los huesos largos (fémur, tibia, húmero, fíbula) o en la pelvis; con menos frecuencia aparece en columna y costillas; el compromiso de manos o pies es raro, y se presenta excepcionalmente a nivel epifisiario.
El diagnóstico se realiza mediante clínica, radiología e histología (biopsia).
| Tumores óseos de células redondas pequeñas | ||||||||||
|
Histopatología:
En la actualidad, los estudios que utilizan marcadores inmunohistoquímicos, citogenéticos, genética molecular y cultivo de tejido indican que estos tumores derivan todos de la misma célula madre primordial, es decir, células pluripotenciales que se pueden diferenciar a células con características mesenquimales, epiteliales, e incluso neurales. La hipótesis más aceptada en la actualidad es que se originan de las células colinérgicas parasimpáticas postganglionares. El sarcoma de Ewing puede considerarse la forma más primitiva ya que está formado por células primitivas y sin diferenciación, mientras que en el PNET sus células poseen una clara diferenciación neural. Entre ambos se encuentra el sarcoma de Ewing atípico que es un estadio intermedio.
Clasificación celular
Los sarcomas de Ewing/PNET son tumores malignos caracterizados histológicamente por células pequeñas redondas, uniformes, empacadas densamente, con núcleo redondo libre de nucléolo y citoplasma indistinto. Tanto los marcadores inmunohistoquímicos (gen mic2) como genéticos (desplazamientos t(11;22) y t(21;22)), así como también la proteína de fusión novel EWS-FLI1 han sido expresados por el sarcoma de Ewing/PNET. Los sarcomas de Ewing/PNET que muestran diferenciación histológica mínima podrían ser confundidos con otros tumores primitivos infantiles de células redondas que comprenden el neuroblastoma, el rabdomiosarcoma, el linfoma y el osteosarcoma de células pequeñas. Los estudios diagnósticos citogenéticos/moleculares, la inmunocitoquímica y la microscopia electrónica podrían ayudar a diferenciar estos tipos de tumores. De acuerdo a los estudios inmunohistoquímicos moleculares, el sarcoma de Ewing/PNET cae dentro del espectro fenotípico de una sola entidad.
Anatomía microscópica
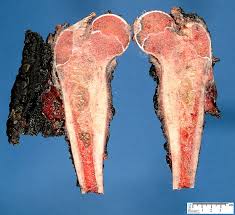
Se caracteriza por una masa densa, difusa, desestructurada y con aspecto homogéneo compuesta por células uniformes de pequeño tamaño con citoplasma claro y núcleos redondos e hipercromáticos, de localización central que al teñirse con hematoxilina le dan un aspecto azulado de ahí el nombre de “sarcomas de células pequeñas redondas y azules” con el que se conoce este grupo de tumores.
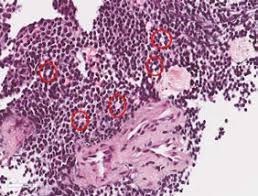
El tumor carece de estroma. Solo se encuentran algunos vaso alrededor de los cuales se disponen los nidos celulares. El tejido tumoral está dividido en lóbulos o hileras por gruesos tabiques de tejido conjuntivo. Como los bordes celulares están poco definidos parece como si los núcleos estuvieran flotando en un mar de citoplasma. Con frecuencia se observan focos de necrosis alrededor de los nidos celulares. Lo básico de este tumor es la ausencia de arquitectura definida, por tanto no se ven las rosetas o pseudorosetas de los tumores de tipo neuroectodérmico.
El citoplasma posee poca cantidad de organelas, sus límites son imprecisos y tiene aspecto vacuolado debido al contenido en glucógeno, que se pone de manifiesto con la tinción de PAS o mejor con la tinción de rojo Best. Las figuras mitóticas son raras.
Como se ha mencionando este tumor no produce matriz, pero puede observarse formación reactiva de hueso que se puede confundir con formación de hueso tumoral y diagnosticarlo como osteosarcoma de células pequeñas. En casos extremadamente raros puede tener una diferenciación epitelial y confundirse con histiocitoma fibroso maligno, osteosarcoma o adamantinoma de huesos largos.
Microscopía electrónica
Muestra unas propiedades ultraestructurales muy característica. Hay dos tipos de células:
-“Células principales”: Son células poligonales, con citoplasma de límites imprecisos, aspecto vacuolado por su contenido en glucógeno, con pocas organelas y frecuentemente un aparato de Golgi prominente. No todas las células contienen glucógeno, y en estos casos su citoplasma adopta una aspecto transparente, por lo que se les conoce como “células claras”. Los núcleos son redondos, pequeños e hipercromáticos, de localización central. Poseen, generalmente, un único nucleolo prominente.
– “Células secundarias oscuras” (por su citoplasma más oscuro). son células del mismo tamaño que las anteriores. Se distribuyen irregularmente con respecto a las principales. Se puede observar apoptosis y condensación cromática. Existe la duda de si estas células son precursoras o formas terminales de las células principales.
Estos dos tipos celulares aparecen entremezclados en los mismos campos y no adoptan ninguna arquitectura definida, por tanto no se ven rosetas o pseudorosetas de Homer-Wrigth. Las células tumorales se agrupan de forma apretada entre si, y crecen siguiendo un patrón difuso sin ningún indicio de organización.
Tumor neuroectodérmico primitivo (PNET) |
El tumor neuroectodérmico primitivo es el último eslabón en la cadena de maduración del sarcoma de Ewing convencional. Se localiza en las vainas nerviosas periféricas.
La apariencia histológica del PNET difiere en cierta forma del sarcoma de Ewing óseo y extraóseo. Estos tumores se componen típicamente de células hipercromáticas que varían de lo redondo a lo oval con citoplasma mínimo. Las células tumorales típicamente se agrupan en nidos y trabéculas formando rosetas variables, y a diferencia de el tipo anterior, destaca la magnitud y la cantidad de las mismas. Las rosetas pueden tener un lumen central, pero con frecuencia son mal definidos y están compuestas por grupos de 6 a 8 células tumorales alrededor de un espacio vacío central. Estás células también contienen glucógeno, red reticular y depósitos de colágeno, PAS positiva. El patrón de crecimiento clásico lobular se aprecia mejor usando un volumen de aumento bajo, y difiere del típico crecimiento difuso de los clásicos de Ewing. Ocasionalmente grupos de células redondas citológicamente uniformes, con cromatina dispersa que semejan a las clásicas de Ewing son vistas intercaladas en lo que de otra forma sería un típico PNET. Esta superposición de características fortalece el concepto de que estos tumores son en realidad el mismo tumor con un espectro de diferenciación. PNETs también muestran variedad de tinción con algunos marcadores neurales incluyendo la enolasa neuroespecífica, Leu-7, sinaptofisin, neurofilamento, y S100. La presencia de gránulos neurosecretores mediante microscopia electrónica, ayuda al patólogo en el diagnóstico de PNET. Estos tumores pueden confundirse con el neuroblastoma del adulto, pero en este hay ausencia de CD99 y alteración del 1p16, cosa que no ocurre en los tumores de la familia del Ewing. En cambió en estos últimos hay una translocación en t(11;22)
Inmunohistoquímica
Si bien no existe ningún hallazgo inmunohistoquímico que permita diferenciar estas neoplasias de otros tumores indiferenciados de la infancia, en la mayoría de los casos los EFT presentan elevados niveles de un antígeno determinado por el gen MIC2, dentro de la región pseudoautosómica de los cromosomas X e Y. El producto de este gen es una glicoproteína de superficie (CD99 o p30/32MIC2) supuestamente involucrada en la adhesión celular. El anticuerpo monoclonal HBA-71, desarrollado contra esta proteína, se muestra positivo en el 95-98% de los casos de los EFT. Aunque la detección inmunohistoquímica de la proteína del MIC2 localizada en la membrana es un parámetro sensible, carece de suficiente especificidad, pero resulta útil en el diagnóstico de estos tumores cuando los resultados se interpretan en el contexto de parámetros clínicos y patológicos. El MIC2 no es un patognomónico del sarcoma de Ewing, además la positividad por inmunohistoquímica se encuentra en otros tumores como el sarcoma sinovial. Durante mucho tiempo el diagnóstico de estos tumores ha sido, por ende, de exclusión. No obstante, en los últimos años se han identificado numerosas alteraciones genéticas específicas para su confirmación diagnóstica.
Patología molecular
Un avance importante fue la identificación de translocaciones específicas en determinados genes que se recombinan para crear productos de fusión. Los genes fusionados expresan y codifican nuevas proteínas que combinan dominios funcionales en moléculas separadas.
Estudios citogenéticos de los EFT han identificado una translocación cromosómica balanceada, t(11;22) (q24;q12). Esta translocación está asociada a una fusión génica aberrante entre el llamado gen EWS, localizado a nivel 22q12 y el gen FLI1 perteneciente a una familia conocida como ETS (Erytroblastic Transforming Sequence) situado en el cromosoma 11q24. A nivel molecular, la porción del cromosoma 22 banda q12, que puede afectar a otros cromosomas, entre los que se incluye el 11 o el 21, se agrupa en el gen EWS mientras que los productos del 11q24 están en el gen FLI1. Característicamente, el amino-terminus del gen EWS está en yuxtaposición con el carboxy-terminus de uno de los otros dos cromosomas. En la mayoría de los casos, (90%) el carboxy-terminus es proporcionado por el gen FLI1 uno de los miembros de la familia de genes de factores de transcripción ETS, localizado en el cromosoma 11 banda q24. Este es usualmente aparente mediante análisis citogenético, el cual revela una desplazamiento entre los cromosomas 11 y 22 en las células malignas.
Esta translocación es un marcador fenotípico de gran importancia. La proteína resultante de la fusión EWS/FLI1 puede ser detectada mediante la técnica de Western-blottin utilizando un anticuerpo policlonal frente a la región c-terminal del FLI1. El 80% de los sarcomas de Ewing expresan esta proteína nuclear, mientras que el neuroblastoma no la expresa. Otros miembros de la familia ETS que podría combinarse con el gen EWS para formar dicho sarcoma, en orden de frecuencia son el ERG (localizado en el cromosoma 21), ETV 1 (localizado en el cromosoma 7), y E1AF (localizado en el cromosoma 17), lo cual resulta en los siguientes desplazamientos: t(21;22), t(7;22), y t(17;22) respectivamente.
Además de estas aberraciones consistentes que comprometen el gen EWS en el 22q12, también se han encontrado aberraciones numéricas y estructurales en los EFT, tales como ganancias en los cromosomas 2,5,7,8,9 y 12 el desplazamiento no recíproco t (1;16) (q12;q11.2) y delecciones en el grupo menor del cromosoma 1. Una prueba molecular ([Retrotranscripción – reacción en cadena de la polimerasa (RT-PCR)] y análisis de restricción de productos (PCR) disponible actualmente solo como instrumento de investigación, ofrece ahora la oportunidad de simplificar de forma destacada la definición de los EFT.
La prueba molecular puede llevarse a cabo en porciones relativamente pequeñas de tejido que se obtienen mediante biopsias mínimamente invasivas, y es capaz de proporcionar resultados más rápidos que el análisis citogenético. Se está evaluando la posible asociación entre los diversos transcritos de fusión y los diferentes comportamientos biológicos de la enfermedad.
La RT-PCR puede diagnosticar micrometástasis ocultas en muestras de sangre periférica o de médula ósea. Así, la detección de la traslocación t(11;22) permite el estudio de la diseminación de la enfermedad y sus factores pronósticos. Por otro lado, como esta traslocación solo está presente en las células tumorales, se podría emplear en el futuro como lesión diana de tratamientos específicos dirigidos a nivel molecular.
Pronóstico:
El pronóstico de estos tumores ha mejorado en gran medida desde el uso de la quimioterapia. Si el tumor está localizado en un área, es menor de 8 cm., y puede ser resecado completamente con la cirugía, el índice de supervivencia a los 5 años es superior al 80%, si se asocia quimioterapia y radioterapia después de la cirugía. Si el tumor no puede ser resecado pero es pequeño, la tasa de supervivencia es superior al 70%. Sin embargo, si el tumor es grande y no se puede resecar completamente, la tasa de supervivencia a los 5 años es menor del 60%, incluso con una buena respuesta a la quimioterapia y radioterapia. En el caso de que haya metástasis en el diagnóstico, la tasa de supervivencia a los 5 años es menor del 30%. Se ha visto que los niños menores de 10 años tienen un mejor pronóstico que los mayores y adolescentes con EFT.
Al diagnóstico, el 30% de los pacientes tienen una diseminación evidente en: El 12% con metástasis en los pulmones, el 12% en los huesos y el 9% en otros sitios del cuerpo (la suma de estos porcentajes es superior al 30% porque algunos pacientes tienen metástasis en más de un área).
Factores pronósticos
-Estructura de la fusión EWS-FLI1: Los pacientes con fusión EWS-FLI 1 tipo 1 presentan mejor comportamiento biológico que los que presentan otros tipos de fusiones, permitiendo una estratificación molecular para tratamiento individualizado. En otros tumores se han identificado perfiles moleculares similares con utilidad diagnóstica y valor pronóstico.
-Sitio: los sitios más favorables son las extremidades distales . Los tumores de localización central (cráneo, clavícula, vértebras y costillas), las extremidades proximales, y la pelvis, tienen peor pronóstico. Los pacientes afectados de tumores primarios distales de las extremidades y tumores primarios que se originan en la clavícula y la escápula han tenido una mejor prognosis que los pacientes con tumores primarios proximales de las extremidades y tumores primarios de las costillas y el área sacropélvica. El Ewing pelviano se descubre típicamente tarde y es, por consiguiente más grande, con un pronóstico más pobre; el índice de supervivencia a los 5 años es aproximadamente 25%.
-Tamaño: El tamaño también influye en el pronóstico; las lesiones más voluminosas tienden a presentarse en los sitios menos favorables.
-Edad: Los niños más jóvenes tienen una supervivencia sin complicaciones mucho mejor que los adolescentes mayores o los adultos.
-Sexo: Las niñas con EFT tienen un mejor pronóstico que los niños.
-Metástasis: Hay estudios que indican que del 50% al 70% de los pacientes sin enfermedad metastática podrían tener una existencia prolongada libre de enfermedad, en comparación con el 20% a 30% de los que presentan enfermedad metastática. Los pacientes con metástasis pulmonar aislada tienen una tasa de supervivencia significativamente más alta que los pacientes con metástasis a otros sitios. Esto es así particularmente en los casos de metástasis pulmonar unilateral.
-Respuesta al tratamiento: Para los pacientes con enfermedad localizada que se hayan sometido a cirugía para extirpar el tumor primario después de recibir quimioterapia neoadyuvante, la respuesta histológica es altamente predictora de los resultados. La respuesta del tumor primario a la quimioterapia fue la variable más importante.
[/vc_column_text][/vc_column][/vc_row][vc_row][vc_column][vc_column_text]
Diagnóstico del diferencial:
Radiológico
En las radiografías simples se observa un patrón apolillado o permeativo que también se ve en otras lesiones: la o steomielitis, granuloma eosinófilo, osteosarcoma osteolítico, neuroblastoma y el linfoma.
-Una lesión grande en mitad de la diáfisis, asociada a tumoración de tejido blando hace pensar en infección o sarcoma de Ewing. La duración de los síntomas pueden ayudar al diagnóstico radiográfico.
-El granuloma eosinófilo: se diferencia por las manifestaciones clínicas.
-El sarcoma de Ewing suele afectar a la diáfisis mientras que el sarcoma osteogénico tiende a ser más metafisario, especialmente en el área de la rodilla. La reacción perióstica es en forma de rayos de sol o triángulo de Codman , cosa que en raras ocasiones ocurre en el sarcoma de Ewing. El osteosarcoma produce, excepto en las formas osteolíticas (telangiectásico y fibroblástico), matriz osteoide que se ve como formación ósea dentro de las lesiones osteolíticas.
-El neuroblastoma metastásico solo se presenta en niños menores de 5 años.
-El linfoma se da en pacientes de más edad y no presenta masa de partes blandas.
-No hay diferencias radiográficas entre el sarcoma de Ewing y el TNEP.
Anatomopatológico
El diagnostico diferencial de los tumores de células pequeñas redondas no es posible si no se recurre a la microscopía electrónica y sobre todo a la inmunohistoquímica.
-Linfoma maligno: los núcleos son mas grandes y menos uniformes. Las células están rodeadas por fibras de reticulina, la tinción de PAS es negativa y el ALC es positivo.
-Linfoma de células grandes: en este, el ALC es positivo, no posee glucógeno intracelular y si que posee fibras de reticulina, a diferencia de los EFT.
-Granuloma eosinófilo: posee células de gran tamaño, de origen histiocitario y que tienen reacción positiva a la proteína S-100
-Osteomielitis crónica: existe abundancia de células plasmáticas o linfocitos. En estos casos , el cuadro histológico, la existencia de granulocitos y la reacción positiva al ALC permiten el diagnóstico diferencial con facilidad.
-Osteosarcoma de células pequeñas (microcítico): para el diagnóstico diferencial nos puede ayudar la presencia de sustancia osteoide y la positividad de los marcadores del sarcoma de Ewing.
-Condrosarcoma mesenquimatoso: en este caso la proteína S-100 es positiva y los marcadores de los EFT son negativos. También puede ayudar la presencia de matriz cartilaginosa.
-Neuroblastoma metastásico. Sus células poseen mayor cantidad de citoplasma y gener almente, se disponen en rosetas, además son positivas para SNE, si bien esta también puede ser positiva en el Ewing atípico y en el TNEP. El TNEP puede presentar células dispuestas en rosetas y presenta también fibrillas, pero estas fibrillas no se ven tan bien perfiladas como en el neuroblastoma.
Presentación clínica:
Estos tumores pueden desarrollarse en casi todos los huesos y tejidos blandos y, por lo general, se presentan como una masa localizada y dolorosa. Puede apreciarse una prominencia ósea, sensible con masa de tejido blando. En otras ocasiones debutan con síntomas y signos generales como fiebre, pérdida del apetito, leucocitosis , aumento de la VSG, aumento de la LDH, y anemia que pueden imitar la osteomielitis. Esta forma de presentación se ha asociado a diseminación y en consecuencia a peor pronóstico. En raras ocasiones puede presentarse con una fractura patológica o con signos de compresión medular o neurológica.
Casi el 25% de los enfermos tiene enfermedad metastásica de pulmón, hueso o médula ósea en el momento del diagnóstico, y casi todos tienen micrometástasis.
Biopsia
Debe seguir los principios básicos.
Se debe obtener un buen fragmento para poder realizar microscopía electrónica y citogenética. El componente de partes blandas es viable y, a menudo proporciona más información que el componente intramedular necrótico. La punción aspiración puede ser suficiente como único método diagnóstico, y es especialmente útil en las localizaciones difíciles.
Estudios de imagen
Radiografías:
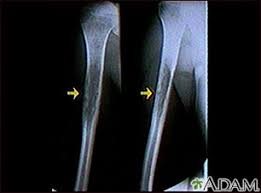
Tumor diafisario mal definido, permeativo o apolillado, con densidad moteada o desigual. La implicación cortical puede producir reacción perióstica en hojas de cebolla y menos frecuente en rayos de sol, asociado a una masa importante de partes blandas, en ocasiones esta es el único hallazgo. En algunos casos esta masa puede producir una erosión cortical en forma de paltillo, que es característica pero no patognomónica, ya que puede aparecer en otros tumores y en la osteomielitis.
El tumor no produce matriz extracelular, pero debido a la gran reacción perióstica que tiene puede confundirse con un osteosarcoma.
Gammagrafía ósea
Hay hipercaptación con el tecnecio. Esta técnica es útil para descubrir metástasis esqueléticas.
En la gammagrafía con el galio se identifica mejor la extensión del tumor a partes blandas. En las lesiones tratadas su captación disminuye más rápidamente que la del tecnecio, y en las recidivas se acumula con mayor velocidad. Es por ello que la gammagrafía con galio puede resultar de utilidad en el seguimiento y en el estudio de las lesiones dudosas tras el tratamiento
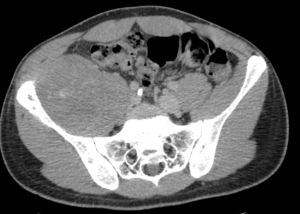
TAC
Proporciona datos acerca de la extensión intramedular . Permite detectar los cambios óseos y corticales mediante la ventana ósea, además de delimitar la afectación extraósea.
Resonancia Magnética
Es la mejor técnica par evaluar la extensión local del tumor. Muestra la extensión extra e intraósea del tumor, así como si el tumor atraviesa la placa epifisaria y la relación con los vasos sanguíneos. Las zonas hipocelulares y las de necrosis tienen menor intensidad de señal. con gadolinio, la hipercaptación se produce solo en las áreas celulares, permitiendo la diferenciación entre tumor y edema peritumoral.
Angiografía
Es útil en la planificación previa a la cirugía conservadora.
TRATAMIENTO
QUIMIOTERAPIA
El Sarcoma de Ewing, como todos los tumores de células redondas, son sensibles a la quimioterapia.
La quimioterapia forma parte de todas las estrategias terapéuticas, tanto en enfermedad local como diseminada.
Quimioterapia adyuvante
Indicada en la enfermedad localizada después de la cirugía. Con la quimioterapia adyuvante se intenta eliminar las micrometástasis.
Quimioterapia neoadyuvante
Con ella se pretende reducir el tamaño del tumor primario, para facilitar la acción de los tratamientos locales. Teóricamente también elimina las micrometástasis. Con este régimen se puede conocer la quimiosensibilidad del tumor de cara a la adyuvancia, pero en contrapartida, si no es eficaz, retrasa el tratamiento local.
Aspectos generales de las opciones de tratamiento
Es de suma importancia para todo paciente, el contar con evaluaciones de diferentes especialistas médicos (p.ej., radiólogo, quimioterapéuta, patólogo, oncólogo cirujano u ortopeda y radiooncólogo) tan pronto como sea posible. El cirujano u ortopeda oncólogo que llevará a cabo finalmente la cirugía, debería participar en la biopsia, para de esa forma poder hacer la incisión en un lugar aceptable. Esto es de particular importancia, si se piensa que la lesión puede ser totalmente extirpada, o si se está pensando en intentar salvar el miembro. El radiólogo y el patólogo especializado en oncología, deben ser consultados previo a la biopsia/cirugía para asegurarse de que el lugar de la incisión no comprometerá el portal de la radiación, y de que se hayan extraído suficientes muestras de tejido (p.ej., cultivos de tejidos para impresiones citogenéticas tejidos congelados, así como tejido fijado en formalina. Se debe de llevar a cabo exámenes de diagnósticos tales como (rayos x del tórax, tomografías computarizadas del tórax RM, o CT del tumor primario) antes de realizar cualquier procedimiento que requiera anestesia, debido a que podría resultar difícil evaluar cualquier anormalidad inducida por la anestesia, que se presente en el tórax. Si el índice de sospecha es alto, antes de realizar la operación se podría llevar a cabo otros exámenes como tomografía del hueso y biopsia de médula ósea.
El tratamiento eficaz de los pacientes con sarcoma de Ewing/EFTs requiere el uso de quimioterapia con múltiples medicamentos, además de radioterapia o terapia quirúrgica al tumor primario. Muchos pacientes con enfermedad metastásica al momento del diagnóstico responden positivamente a la terapia administrada a pacientes con enfermedad localizada; sin embargo, en la mayoría de los casos la enfermedad solo logra controlarse de forma parcial, o recurre. Aquellos pacientes que solo presentan metástasis del pulmón tienen una mejor sobrevivencia sin complicaciones que aquellos con metástasis al hueso o a la médula ósea.
Algunos grupos pequeños no aleatorios con alto riesgo de recaída, indicaron una mejoría en la supervivencia mediante el uso de dosis muy altas de quimioterapia con radioterapia y posiblemente cirugía con o sin trasplante autólogo de médula ósea. En un estudio mayor donde se incluyó a todos los pacientes desde el diagnóstico inicial, no se pudo mostrar el beneficio de una terapia de alta dosis con reconstitución autóloga de células madre utilizada como terapia de consolidación. El riesgo de una leucemia secundaria puede aumentar con tratamientos de dosis intensivas.
Los pacientes en tratamiento de EFTs, corren un riesgo significativamente mayor de desarrollar malignidades secundarias que la población general.
TRATAMIENTO ESTANDAR
Puesto que casi todos los pacientes con enfermedad aparentemente localizada en el momento del diagnóstico tienen enfermedad metastásica oculta, el tratamiento indicado para todos los pacientes es quimioterapia con múltiples fármacos a la vez que se controla la enfermedad mediante cirugía, radiación o ambas.
Los estándares quimioterapéuticos actuales en los Estados Unidos incluyen la vincristina, doxorrubicina, y ciclofosfamida (VAdriaC) alternando con ifosfamida y etopósido. La importancia de la doxorrubicina ha sido demostrada en ensayos comparativos aleatorios. El aumento en la intensidad de la dosis de doxorrubicina durante los primeros meses de terapia que dieron como resultado una supervivencia sin eventualidades. Estos resultados han llevado a descontinuar el uso de la actinomicina-D en protocolos intergrupales. La combinación de ifosfamida y etopósido han demostrado actuar en los tumores óseos de Ewing (ETB), y un estudio clínico de magnitud, aleatorio y un ensayo no aleatorio, demostró que los resultados mejoraban cuando la combinación de ifosfamida y etopósido se administraba en combinación con cursos alternos de VAdriaC. La utilización de dosis altas de VAdriaC ha dado resultados positivos en un número limitado de pacientes. Sin embargo, aún no se ha probado la validez de una terapia de dosis intensiva en los pacientes con sarcoma de Ewing localizado, y podría estar relacionado con un aumento del riesgo de desarrollar una malignidad secundaria.
El control local puede lograrse mediante cirugía, radiación o ambas. La cirugía suele ser el enfoque de elección siempre que la lesión sea resecable. Nunca ha podido probarse la superioridad de la resección local para el control local durante un estudio aleatorio prospectivo. La aparente superioridad podría representar una parcialización en la selección. En estudios anteriores, los tumores mas pequeños y periféricos tuvieron mas probabilidades de ser tratados con cirugía, mientras que los tumores mayores y mas centrales tuvieron la probabilidad de ser tratados con radioterapia. Si un niño muy joven padece de ETB, la cirugía podría ser una terapia menos mórbida que la radioterapia debido a que la radiación retrasa el crecimiento óseo. Otro de los beneficios potenciales de la resección quirúrgica del tumor primario, lo es la información relacionada con la cantidad de necrosis del tumor resecado. Los pacientes con tumor residual viable en el espécimen resecado, tiene un resultado más precario cuando se compara con aquellos que exhiben una necrosis completa. En el estudio francés sobre Ewing (EW88), la supervivencia libre de complicaciones en aquellos pacientes con menos de 5% de tumor viable, los que tenía de un 5% a 30% de tumor viable y los de más de 30% de tumor viable fue de 75%, 48% y 20% respectivamente. Al presente existen estudios europeos en curso que investigan si la intensificación del tratamiento ()quimioterapia de alta dosis con rescate de células madres [SCR, siglas en inglés] mejorará los resultados en pacientes con respuesta histológica precaria. La radioterapia se emplea en pacientes que no tienen la opción de una cirugía que preserve la funcionalidad y en pacientes que cuyos tumores han sido resecado pero con márgenes inadecuados.[/vc_column_text][vc_column_text]La radioterapia debe ser llevada a cabo en un ambiente en el que se aplican estrictas técnicas de planificación por profesionales experimentados en el tratamiento de tumores del grupo de Ewing. Tal enfoque resultará en un control local del tumor con morbilidad aceptable en la mayoría de los pacientes. La dosis de radiación puede ser ajustada dependiendo de la extensión de la enfermedad residual después del procedimiento quirúrgico. La radioterapia se administra generalmente en dosis de 5600 cGy a la extensión tumoral prequimioterapia. Un estudio aleatorio con 40 pacientes con ETB en el que se uso 5580 cGy en una extensión tumoral con un margen de 2cm prequimioterápica comparada con la misma dosis tumoral total seguido de 3960 cGy al hueso entero, no mostró ninguna diferencia en cuanto a control local o supervivencia libre de enfermedades. La radioterapia hiperfraccionada no estuvo asociada en ningún momento con un control local, o una disminución en la morbilidad. Algunos pacientes podrían requerir resección quirúrgica después de la radioterapia.
Grupo de tumores metastásicos de Ewing
El pronóstico de pacientes con enfermedad metastásica es precario.
Opciones de tratamiento estándar
Tratamiento estándar alternando vincristina, doxorrubicina, ciclofosfamida, e ifosfamida/etopósido combinado con radioterapia a todos los sitios de la enfermedad macroscópica y posiblemente la excisión quirúrgica selecta para el sarcoma de Ewing/PNET metastásico con frecuencia resulta en respuestas parciales o completas; sin embargo la tasa de curación es del 20%. En los pacientes con metástasis pulmonar/pleural únicamente, la tasa de curación es de aproximadamente 30%. Los pacientes que no recibieron irradiación de los pulmones obtuvieron peores resultados que los que la recibieron. Los pacientes con metástasis ósea o de la médula ósea únicamente tienen aproximadamente una tasa de curación del 20 al 25%. Los pacientes que sufren de una combinación metastásica de pulmón y hueso/médula ósea tienen una tasa de curación de menos del 15%.
La radioterapia deberá administrarse en un ambiente en el cual se aplican técnicas estrictas de planificación por quienes son expertos en el tratamiento del sarcoma de Ewing/PNET. Dicha estrategia dará lugar a un control local del tumor con una morbilidad aceptable en la mayoría de los pacientes. Se deberá considerar la administración de radioterapia al tumor primario así como también a los sitios de enfermedad metastásica, pero interferir con la administración de quimioterapia si se incluye demasiada médula ósea en el campo. Los sitios metastásicos de enfermedad en el hueso y los tejidos blandos pueden recibir radioterapia de 4500 a 5600 cgy. Todos los pacientes con metástasis pulmonar deben someterse a radiación pulmonar completa, aun cuando se haya obtenido una resolución completa de la metástasis pulmonar con la quimioterapia. Las dosis de radiación son moduladas de acuerdo al tamaño de la porción del pulmón que será irradiada. Se usan dosis de 1200 a 1500 cgy cuando se va a tratar todo el pulmón.
Existen terapias más intensivas, muchas de las cuales incorporan quimioterapia de alta dosis con o sin irradiación total del cuerpo junto con soporte de células madres, estas, sin embargo, no han logrado mostrar mejoría en cuanto a la tasa de supervivencia sin complicaciones en pacientes con metástasis al hueso o la médula ósea. En este momento se desconoce el impacto de una quimioterapia con dosificación alta junto con el apoyo de las células madres periféricas sanguíneas en pacientes con metástasis pulmonar.
Escisión quirúrgica:
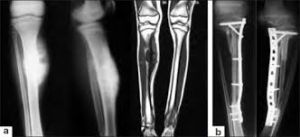
Su finalidad es eliminar la enfermedad local. Actualmente se prefiere la cirugía conservadora, sobre todo en las extremidades. No obstante se puede recurrir a la amputación en casos de recidivas en una extremidad irradiada previamente, y sobre todo si ya se había aplicado cirugía conservadora, algunos casos de fracturas patológicas al diagnóstico, tumores grandes que no responden a la quimioterapia neoadyuvante y técnicamente sean difíciles de resecar.
En los niños se pueden salvar los miembros mediante la colocación de prótesis expandibles.
Hay estudios que indican que la cirugía es superior a la radioterapia en términos de control local del tumor
La cirugía es preferida sobre la radioterapia si:
– el hueso implicado es prescindible (ejemplo: peroné, costilla, clavícula);
– si la radioterapia dañara el platillo de crecimiento (la radioterapia puede causar cierre prematuro de platillo de crecimiento);
– si hay fractura patológica;
