Ortopédica Mexicana 2009; 23(6): Nov.-Dic: 376-382
Sansón-RíoFrío JA, Navarro Santiesteban S, Bahena RI, Villavicencio VV, Martínez-Said H,* Padilla RA, Cuellar HM[Las encondromatosis comprenden un grupo heterogéneo de síndromes congénitos caracterizados por la presencia de encondromas múltiples, asociados a malformaciones musculoesqueléticas secundarias al acortamiento de las extremidades, escoliosis, fracturas en terreno patológico y seudoartrosis. La principal complicación de los encondromas es su transformación maligna a condrosarcomas, que puede llegar a presentarse hasta en el 25% de los casos. Los múltiples síndromes encondromatosos presentan muchas similitudes clínicas, por lo que es difícil realizar un diagnóstico diferencial. Exponemos el caso clínico de un paciente de 38 años con diagnóstico de encondromatosis múltiple familiar, el cual desarrolló un condrosarcoma pélvico, tratado con hemipelvectomía externa. Revisamos la literatura referente a los aspectos específicos de los síndromes de Mafucci, Ollier y encondromatosis múltiple familiar.Las encondromatosis son un grupo de patologías caracterizadas por la presencia de lesiones discondrodisplásicas localizadas en las metáfisis y diáfisis de los huesos en crecimiento, principalmente de las extremidades, no obstante también se han reportado en sitios menos habituales como son: pelvis, costillas, base del cráneo, septum nasal, senos paranasales y tráquea.1-4 Dichas lesiones son generalmente benignas, sin embargo su principal complicación es la transformación maligna en condrosarcomas. La supervivencia a 10 años de los pacientes con condrosarcomas de grado histológico I es hasta del 80%, sin embargo, esta sobrevida disminuye hasta un 31% en tumores de grado histológico III.
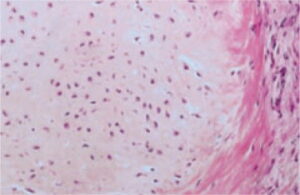
(Imagen Histologica)
La cirugía es la piedra angular del tratamiento debido a la resistencia de los encondromas a la radio y quimioterapia.5 Reportamos el caso de un paciente con encondromatosis múltiple familiar que presentó un condrosarcoma en la pelvis y fue tratado con hemipelvectomía externa. Así mismo se presenta una revisión de la literatura de las encondromatosis y su asociación con el condrosarcoma.
La enfermedad de Ollier y el síndrome de Maffucci son las encondromatosis mejor descritas y estudiadas en la literatura, sin embargo no son las únicas, existe una gran variedad de estos síndromes, entre los que también destaca la exostosis múltiple familiar.1,6 La primera de estas patologías fue descrita en el año de 1900 por el cirujano francés Leopold Ollier, como una enfermedad no hereditaria, de etiología desconocida, sin predominio de género, que se presenta entre el segundo y décimo año de vida.2 Se caracteriza por la presencia unilateral de discondroplasia y múltiples encondromas, localizados en los huesos largos de las extremidades7 y asociados a anormalidades

esqueléticas, secundarias al acortamiento de las extremidades, liosis, fracturas en terreno patológico y seudoartrosis.5,8 Cuando además de los datos ya mencionados se encuentran hemangiomas viscerales o mucocutáneos se conoce como síndrome de Maffucci, nombrado así por el patólogo italiano Angelo Maffucci, quien lo describió en el año de 1881. En este síndrome los mas son más frecuentes en los huesos cortos de las manos y pies y son diagnosticados durante la edad preescolar.3,5,6 El diagnóstico diferencial de estas dos entidades es difícil, debido a sus similitudes, por lo que se ha propuesto que son diferentes grados de expresión de una misma patología.2,9 El origen de estas enfermedades aún es desconocido, pero se cree que se debe a una displasia mesodérmica congénita o a una anomalía cromosómica aún no identificada.10,11 Una tercera entidad es la exostosis múltiple, la cual, a diferencia de los síndromes anteriores, tiene un patrón de herencia ya conocido: autosómico dominante. Se han identificado dos genes supresores tumorales, involucrados en esta enfermedad: EXT1 y EXT2, localizados en los cromosomas 8q24 y 11p11-p12, respectivamente. Actualmente se sugiere la existencia de un tercer gen (EXT3) en el cromosoma 19p, sin embargo aún no ha sido identificado.12-14 Su cuadro clínico se caracteriza por el desarrollo de dos o más encondromas en las epífisis de los huesos largos, principalmente en la articulación femorotibial. Se presenta durante la primera década de la vida, con una edad media de diagnóstico a los 3 años de edad. A diferencia de otras encondromatosis, una vez que se cierran los cartílagos de crecimiento, se detiene la formación de nuevos encondromas.15 Las malformaciones musculoesqueléticas más frecuentes son: el acortamiento del cúbito con arqueamiento del radio (39-60%), crecimiento asimétrico de las extremidades (10-50%), baja estatura (37-44%), angulación en varo o valgo de la rodilla (8-33%) y deformidad de tobillo (2-54%).14 El condrosarcoma representa 1-11.4% de las lesiones óseas malignas y se origina en el 80% de los casos a partir de un osteocondroma, aunque existen otras lesiones cartilaginosas precursoras como son: la condromatosis sinovial, displasia fibrosa, exostosis cartilaginosa y lesiones radioinducidas. En el caso de los encondromas que se presentan de forma aislada, el riesgo de transformación a condrosarcoma es menor al uno por ciento.1,3 Sin embargo, puede llegar a ser hasta de 25% en los pacientes mayores de 40 años con enfermedad de Ollier y hasta de 100% para los pacientes con síndrome de Maffucci.3 Los condrosarcomas se clasifican en primario o secundario, en relación a la presencia de lesiones precursoras, de acuerdo a su topografía en central o periférico y por grado histológico en bien, moderadamente o pobremente diferenciados. Esta última clasificación es la más utilizada, ya que ofrece un valor pronóstico derivado del comportamiento biológico del tumor. El sitio más común de metástasis son los pulmones. Los pacientes con síndrome de Maffucci generalmente desarrollan condrosarcomas de grado III, lo cual les confiere un pobre pronóstico, mientras que los pacientes con enfermedad de Ollier presentan condrosarcomas de bajo grado.3 Ambos síndromes se han asociado a neoplasias benignas y malignas como son: los adenomas tiroideos, paratiroideos e hipofisiarios, así como astrocitomas, hemangiosarcomas, linfangiosarcomas, cáncer de ovario y páncreas.
PRONOSTICO
El pronóstico de los pacientes empeora cuando se presenta alguno de estos tumores.2,15 El diagnóstico de las encondromatosis se realiza bajo la sospecha clínica y 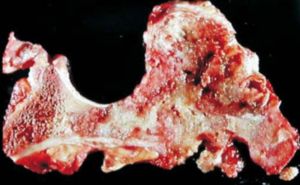 se apoya en
se apoya en
estudios de imagen. No existen estudios específicos para su diagnóstico.5 Los pacientes parecen normales al nacimiento, sin embargo, durante su infancia presentan deformidades esqueléticas secundarias al desarrollo de los encondromas o la presencia de fracturas en terreno patológico.16 Los hallazgos clínicos más comunes son: acortamiento de las extremidades, lesiones tumorales y fracturas de las áreas afectadas, la enfermedad puede progresar hasta la discapacidad del paciente o autolimitarse al final de la adolescencia.10 Los datos sugestivos de malignidad de un encondroma son dolor y crecimiento progresivo.16 La radiografía simple es el estudio inicial en la evaluación de los encondromas que generalmente se encuentran localizados en la diáfisis de los huesos largos y se observan como lesiones ovaladas, lobuladas y bien delimitadas.3 La TAC permite valorar la formación de matriz ósea y los órganos intraabdominales, mientras que la RMN valora la extensión a tejidos blandos, permitiendo la identificación de hemangiomas y tumores en el SNC.2-4 Una vez identificado un encondroma con sospecha de malignidad, lo recomendado es tomar una 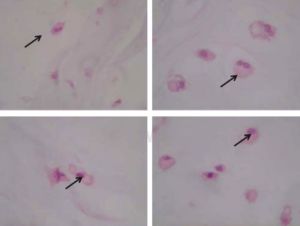 biopsia trucut guiada por imagen.16 Desde el punto de vista histopatológico, el diagnóstico diferencial de los encondromas y los condrosarcomas de bajo grado es difícil, ya que ambas entidades presentan hipercelularidad y atipia leve, por lo que el diagnóstico diferencial se apoya en la agresividad del cuadro clínico y en los estudios de imagen.2,16
biopsia trucut guiada por imagen.16 Desde el punto de vista histopatológico, el diagnóstico diferencial de los encondromas y los condrosarcomas de bajo grado es difícil, ya que ambas entidades presentan hipercelularidad y atipia leve, por lo que el diagnóstico diferencial se apoya en la agresividad del cuadro clínico y en los estudios de imagen.2,16
La cirugía es la piedra angular del tratamiento de los condrosarcomas, debido a su mala respuesta al tratamiento con quimioterapia y radioterapia. El objetivo del tratamiento quirúrgico es obtener márgenes libres de tumor, conservando la función y el aspecto cosmético de los órganos involucrados mediante una resección amplia.
Conclusiones
El diagnóstico de las encondromatosis se establece por la sospecha clínica y se apoya en los estudios de imagen.3 Dado que estas patologías comparten muchas características clínicas, es difícil establecer un diagnóstico diferencial, el antecedente de familiares afectados orienta hacia la encondromatosis múltiple familiar, en tanto que la localización unilateral sugiere enfermedad de Ollier y la presencia de encondromas bilaterales orienta hacia el síndrome de Mafucci. La determinación genética de los genes EXT1 y EXT2 es el estudio más específico para el diagnóstico de la encondromatosis múltiple familiar, sin embargo, no en todos los hospitales se puede realizar este estudio (Tabla 1). 13 La radiografía simple se utiliza en la evaluación inicial de los condrosarcomas secundarios, las lesiones típicas son ovaladas, lobuladas, largas y bien delimitadas, generalmente en la diáfisis de los huesos afectados.3
El crecimiento o dolor de un encondroma es sugestivo de malignidad.13 Ante la sospecha de transformación maligna se debe realizar una biopsia guiada por TAC o Intensificador de imagen.16 En el caso de la EHM se puede realizar una resonancia magnética en T2, donde el engrosamiento mayor a 1 cm de la capa cartilaginosa de las 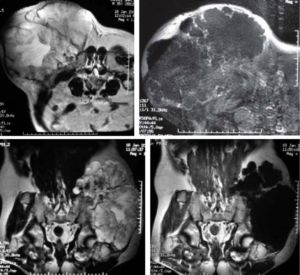 epífisis es sugestivo de malignización.13,14 Los encondromas tienen un riesgo mayor de malignidad (25%) en comparación a cuando se presentan de forma aislada3 y requieren tratamiento sólo en el caso de sintomatología o sospecha de malignidad, en cuyo caso el tratamiento de elección es la resección quirúrgica de la lesión. Los condrosarcomas requieren de resección en bloque de la lesión y su pseudocápsula, buscando dejar márgenes libres de neoplasia.14 La recurrencia local de condrosarcomas cuando se realiza resección con márgenes libres de neoplasia es de 3%, sin embargo se puede elevar hasta 23% si los márgenes no se encuentran libres de neoplasia.14
epífisis es sugestivo de malignización.13,14 Los encondromas tienen un riesgo mayor de malignidad (25%) en comparación a cuando se presentan de forma aislada3 y requieren tratamiento sólo en el caso de sintomatología o sospecha de malignidad, en cuyo caso el tratamiento de elección es la resección quirúrgica de la lesión. Los condrosarcomas requieren de resección en bloque de la lesión y su pseudocápsula, buscando dejar márgenes libres de neoplasia.14 La recurrencia local de condrosarcomas cuando se realiza resección con márgenes libres de neoplasia es de 3%, sin embargo se puede elevar hasta 23% si los márgenes no se encuentran libres de neoplasia.14
El grado histológico es el principal factor pronóstico para la supervivencia en los pacientes portadores de condrosarcoma, sin embargo, estos pacientes pueden desarrollar más de un condrosarcoma durante su vida, por lo que se recomienda un seguimiento estrecho. Ante la presencia de nuevas lesiones acompañadas de signos y/o síntomas sospechosos de malignidad, se debe descartar la presencia de condrosarcoma por biopsia.
[/vc_column_text][vc_single_image image=”2487″ img_size=”full”][/vc_column][/vc_row][vc_row][vc_column][/vc_column][/vc_row][vc_row][vc_column width=”1/2″][vc_column_text]1. Rozeman LB, Sangiorgi L, Briare-de-Brujin IH, Mainil-Varlet P, Bertoni F, Cleton-Jansen AM, Hogendoorn PC, Boveé JV: Enchondromatosis (Ollier Disease, Maffucci Syndrome) is not caused by the PTHR1 mutation p.R150C. Hum Mutat 2004; 24(6): 466-73.
2. Ahmed SK, Lee WC, Irving RM, Walsh AR: Is Ollier’s disease an understaging of Maffucci’s syndrome? J Laryngol Otol 1999; 113(9): 861-4.
3. Ramirez J, Padilla A, Romero A, Lavín A, Medina J, Dubón E, Turcios E: Síndrome de Maffucci: Informe de dos pacientes y revisión de la literatura. Cir Ciruj 2005; 73: 217-21.
4. Desai S, Kubeyinje EP, Belagavi CS, Desai S: Maffucci’s syndrome. Ann Saudi Med 1997; 17(4): 451-3.
5. Camarda R, Wilcox III RB, Ghazinouri R, Wilmarth MA: Acute physical therapy management for a patient with Maffucci’s syndrome following modified internal hemipelvectomy and reconstruction with Saddle prosthesis: A case report. Rehab Oncol 2008; 26(1): 8-17.
6. Gabos PG, Bowen JR: Epiphyseal-metaphyseal enchondromatosis. A new clinical entity. J Bone Joint Surg Am 1998; 80(6): 782-92.
7. Pacheco RC, Torres B, Ugalde A, del Vecchyo C, Sastré N: Enfermedad de Ollier de presentación bilateral. Reporte de un caso y revisión de la literatura. Rev Med Hosp Gen Mex 2001; 64(3): 152-6.
8. Pallotta R, Fusilli P, Saponari A: Maffucci’s syndrome. Ital J Pediatr 2003; 29: 437-8.
9. McDermott A-L, Dutt S, Chavda SV: Maffucci’s syndrome: clinical and radiological features of a rare condition. J Laryngol Otol 2001; 115: 845-7
10. Dini LI, Isolan GR, Saraiva GA, Dini SA, Gallo P: Maffucci’s syndrome complicated by intracranial chondrosarcoma: two new illustrative cases. Arq Neuropsiquiatr 2007; 65(3B): 816-21.
11. Hopyan S, Gokgoz N, Poon R, Gensure RC, Yu C, Cole WG, Bell RS, Jüppner H, Andrulis IL, Wunder JS, Alman BA: A mutant PTH/PTHrP type I receptor in enchondromatosis. Nature Genetics 2002; 30.
12. Lopes A, Morini S, Vieira L, Talvane A: Chondrosarcoma secondary to hereditary multiple exostosis treated by extended internal hemipelvectomy. São Paulo Medical Journal 1997; 115(3): 1440-3.
13. Kivioja A, Ervasti H, Kinnunen J, Kaitila I, Wolf M, Böhling T. Chondrosarcoma un a family with multiple hereditary exostoses. The Journal of Bone and Joint Surgery. 2000; 82(2): 261-6.
14. Bovée JVMG: Multiple osteochondromas. Orphanet Journal of rare diseases. 2008; 3: 3.
15. Kobayashi T, Asakawa H, Ogawa A, Kaneko K, Nakano Y, Tamaki Y, Morimoto S, Monden M: Hyperparathyroidism in a patient with Maffucci’s syndrome: A case report. Ann Saudi Med 1997; 17(4): 457-9.
16. Silve C, Jüppner H: Review Ollier disease. Orphanet J Rare Dis 2006, 22: 1:37.
17. Miyake M, Tateishi U, Maeda T, Arai Y, Hasegawa T, Sugimura K: MR Features of angiosarcoma in patient with Maffucci’s syndrome. Rad Med 2005; 23(7): 508-512. Unni KK: Cartilaginous lesions of bone. J Orthop Sci 2001; 6: 457-72.
18. Maffucci A: Di un caso di encondroma ed angioma multipli. Contribuzione alla genesi embrionale dei tumor. Mov Med Chir 1981; 3: 399-412/565-75.[/vc_column_text][/vc_column][vc_column width=”1/2″][vc_column_text]19. Maffucci A: A case of enchondroma and multiple hemangioma [in Ital-ian]. Med Chir Napoli 1881; 3: 399-412. 20. Ollier L: Dyschondroplasia[in Italian]. Bull Soc Lyon Med 1899; 93: 23-4. 21. McDermott A-L, Dutt S, Chavda SV: Maffucci’s syndrome: clinical and radiological features of a rare condition. J Laryngol Otol 2001; 115: 845-7.
22. Strang C, Rannie I: Dyschondroplasia with haemangiomata (Maffucci, syndrome). J Bone Joint Surg 1950; 32: 376-83.
23. Ahmed AR, Tan TS, Unni KK, Collins MS, Wenger DE, Sim FH: Secondary chondrosarcoma in osteochondroma: report of 107 patients. Clin Orthop Relat Res 2003; 1(411): 193-206.
24. Johnson JL, Webster JR, Sippy HI: Maffucci’s syndrome (dyschondroplasia with hemangiomas). Am J Med 1960; 28(5): 864-6.
25. Sun TC, Swee RG, Shives TC, Unni KK: Chondrosarcoma in Maffucci´s syndrome. J bone Join Surg Am 1985; 67: 1214-9.
26. Hunter D, Wiles P: Dischondroplasia (Ollier’s disease). Br J Surg 1935; 22: 507-19.
27. Ozisik YY, Meloni AM, Spanier SS, Busch CH, Kingsley KL, Sandberg AA: Deletion lp in a low-grade chondrosarcoma in a patient with Ollier’s disease. Cancer Genet Cytogenet 1998; 105: 128-33.
28. Collins PS, Han W, Williams LR, et al: Maffucci´s syndrome (hemangiomatosis osteolytica): a report of four cases. J Vasc Surg 1992; 16: 365-71.
29. Pring M, Weber K, Unni K, Sim F: Chondrosarcoma of the pelvis: a review of sixty-four cases. J Bone Joint Surg Am 2001; 83-A: 1630-42.
30. Wirbel R, Schulte M, Mutschler W: Surgical treatment of pelvic sarcomas: oncologic and functional outcome. Clin Orthop Relat Res 2001: 190-205.
31. Ham SJ, Schraffordt Koops H, Veth RP, et al: External and internal hemipelvectomy for sarcomas of the pelvic girdle: consequences of limb-salvage treatment. Eur J Surg Oncol 1997; 23: 540-6.
32. Little C: Ollier disease: an interdisciplinary approach. Orthop Nurs 1994; 13: 50-5.
33. Mellon CD, Carter JE, Owen DB: Ollier´s disease and Maffucci´s syndrome: distinct entities or a continuum. Case report: enchondromatosis complicated by an intracranial glioma. Journal of Neurology 235: 376-8.
34. Schwartz HS, Zimmerman NB, Simon MA, Wroble, RR, Millar, EA, Bonfiglio M: The malignant potential of enchondromatosis. Journal of Bone and Joint Surgery 69: 269-74.
35. Cleaveland M, Fleiding JW: Chondrodysplasia (Ollier´s disease). Report of a case with a thirty-eigth year follow up. J Bone and Joint Surg 1959; 41-A: 1341-4. 36. Fairbank HA: Dyschondroplasia. Synonyms Ollier´s disease multiple enchondromata. J Bone and Joint Surg 1948; 30- B(4): 689-708.[/vc_column_text][/vc_column][/vc_row]
