El condrosarcoma es un tumor maligno de células productoras de cartílago. Es el segundo tumor óseo maligno en orden de frecuencia, y supone el 10-20% de todos los tumores óseos. Pueden ser primarios (80% de los casos) o secundarios que resultan, en  su mayor parte, de la malignización de tumores benignos preexistentes. Lo más frecuente es que se presente como una lesión de bajo grado intracompartimental. Son lentos para metastatizar y pueden crecer hasta inmensas proporciones, tienden a destruir el hueso y extenderse en los tejidos blandos
su mayor parte, de la malignización de tumores benignos preexistentes. Lo más frecuente es que se presente como una lesión de bajo grado intracompartimental. Son lentos para metastatizar y pueden crecer hasta inmensas proporciones, tienden a destruir el hueso y extenderse en los tejidos blandos
La proporción de supervivencia a largo plazo de pacientes tratados de condrosarcoma está entre 50-75%.
La forma primaria ocurre más a menudo en los hombres entre 50-60 años de edad (mientras que el osteosarcoma ocurre entre 20-30 años de edad).
Pronóstico
El pronóstico es variable y depende de la agresividad biológica del tumor. El tratamiento debe basarse en un diagnóstico precoz y exacto, con una biopsia generosa, correctamente planeada y ejecutada, la determinación precisa de la localización del tumor y su resección completa con márgenes amplios con un manguito de tejido normal. La quimioterapia y la radioterapia no son muy eficaces en estos tumores.
Los factores pronósticos más importantes son:
 El tipo de cirugía (márgenes). Se ha demostrado que la incidencia de metástasis, recurrencia local y muerte secundaria al tumor es significativamente más elevada en los pacientes con incisión marginal o incompleta del tumor.
El tipo de cirugía (márgenes). Se ha demostrado que la incidencia de metástasis, recurrencia local y muerte secundaria al tumor es significativamente más elevada en los pacientes con incisión marginal o incompleta del tumor.
Localización. Las lesiones localizadas en la pelvis son de peor pronóstico por diagnóstico más tardío y resección más dificultosa.
El grado histológico del tumor. Es un factor predictivo importante en el comportamiento del tumor, si bien es difícil diferenciar, en ocasiones , tumores de alto y bajo grado, incluso con muestras abundantes.El tipo de cirugía (márgenes).
Se ha demostrado que la incidencia de metástasis, recurrencia local y muerte secundaria al tumor es significativamente más elevada en los pacientes con incisión marginal o incompleta del tumor.La determinación del grado histológico es subjetiva, por lo que se han intentado encontrar métodos objetivos para poder establecer el pronóstico con más fiabilidad. Estos métodos están basados en la investigación de la síntesis y el contenido de ADN, citometría de flujo, marcadores moleculares (p53, MIB-1, src o ERB-1), proliferación celular, citogenética, histomorfometría y clasificaciones radiográficas. Así Lee et al. observaron una relación significativa entre contenido en ADN, aneuploidía y tasas de metástasis y muerte secundaria a la enfermedad
Localización:
La pelvis, el fémur proximal, y la cintura escapular (mientras que el osteosarcoma ocurre más a menudo alrededor de la rodilla). Cuando el condrosarcoma ocurre en la pelvis, es a menudo de alto grado, diagnosticó tardío, y tiene un pronóstico pobre.
El condrosarcoma secundario ocurre en aproximadamente 25% de los casos.
Es una transformación maligna de un encondroma u osteocondroma preexistente.
Variantes de Condrosarcoma:
El grado histológico:De estas clasificaciones la que mayor utilidad tiene en cuanto el pronóstico es la basada en el grado histológico. El sistema de clasificación histológica se divide en tres grados según la celularidad, la atipia y el pleomorfismo.
- Bajo (grado 1), La graduación se basa en la apariencia histológica del tumor; los tumores que se parecen mucho al cartílago normal son de bajo grado (grado1), y tienen el menor riesgo de producir metástasis, su crecimiento es lento y se asocian con una supervivencia de más del 90% a los cinco años.
- Medio (grado 2) Este y el siguiente tienen un menor parecido al cartílago normal, se comportan más agresivamente y tienen mayor potencial de dar metástasis.
- Alto (grado 3). Tienen un riesgo más alto de dar metástasis y es, probablemente el único que llega a metastatizar. La tasa de supervivencia de este tipo y del anterior, a los 5 años, se sitúa entre el 40-50%. Un tipo de condrosarcoma que tiene un riesgo más alto de la metástasis que un condrosarcoma grados 3 es el llamado condrosarcoma desdiferenciado. Se trata de un tipo raro que se genera en una lesión benigna de cartílago o de un condrosarcoma de bajo grado. Tiene un mal pronóstico, ya que la tasa de supervivencia a los 5 años es del 20-30%.
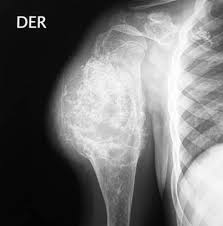
Hay unos subtipos histológicos:
Condrosarcoma de células claras:
Se trata de un condrosarcoma secundarios que nace de una exostosis, solitaria o múltiple con predominio en el sexo masculino y que siempre será después de la pubertad. Tienen un buen pronóstico, mejor que el de otros condrosarcomas y raramente da metástasis
Tienen un buen pronóstico, mejor que el de otros condrosarcomas y raramente da metástasis
Se localiza preferentemente en la pelvis, fémur proximal, columna vertebral y sacro, húmero proximal, costillas, omóplato y fémur distal. Es raro observarlo en la rodilla, por otro lado lugar frecuente de exostosis. En la pelvis se origina más frecuentemente en el ala y días como en el marco anterior. En las vértebras y desacuerdo en el arco posterior. En los huesos largos, se origina siempre en una extremidad diafisarias próxima a la metáfisis. El tumor se extiende por el exterior del hueso, invaden la en exostosis preexistente y, en las formas más agresivas y más avanzadas, también del hueso de implantación.
El signo más importante es la presencia de una masa adherida al hueso y de consistencia y dura, abollonada, de contornos nítidos y que se desarrolla con lentitud. En algún caso el enfermo sufre una exostosis múltiple, en otros casos, estaría perfecto de una exostosis solitario, pero lo más habitual es no encontrar en la anamnesis ningún rastro de exostosis preexistente. La masa que es indolora y en algún caso puede alcanzar dimensiones gigantescas.
Radiográficamente la imagen típica está formada por núcleos opacos debidos a calcificaciones y osificaciones del tejido neoplásico. Las calcificaciones representan en forma de gránulos, anillos o de ” palomitas de maíz”. Las osificaciones están más que estructuradas y forman “sarmientos” culpa indios, imágenes de tabiques en “migas de pan” grumos de opacidad intensa. Se presenta por lo tanto una masa extraóseo única lobulada, abollonada en “coliflor” e intensamente y opaca a los rayos X.. La capa superficial del tumor, de formación más reciente, están menos calcificado y, por consiguiente, puede presentar contornos vagos hacia las partes blandas. Con la ayuda del TAC y la RM puede observarse la parte cartilaginosa no calcificada del tumor. Esta parte cartilaginosa puede representar la mayor parte de la neoplasia. Estas técnicas de diagnóstico puede asimismo demostrara que se trata del primer estadio del paso de la exostosis han condrosarcoma periférico, haciendo visible un aumento del espesor del capuchón cartilaginosa de una exostosis que en la radiografía parece más o menos típica. Asimismo estas técnicas son las únicas que pueden permitir detectar la presencia de nódulos cartilaginosos de recidiva local de tumor en las partes blandas tras una exéresis quirúrgica y inadecuada. El condrosarcoma periférico muestra una importante hipercaptación en la gammagrafía, generalmente más que la de la exostosis. Cuando el condrosarcoma periférico en vuelve al hueso de origen, habiendo invadido y borrado completamente la exostosis, el tronco de implantación es irreconocible y se ve como, en ocasiones, el tumor y invade el tejido esponjoso de hueso contiguo.
Macroscópicamente, cuando en un adulto, la envoltura cartilaginosa de una exostosis alcanza o sobrepasa 1 o 2 cm de espesor, hay que sospechar una malignización (en el niño, el espesor puede alcanzar los 2 cm sin que por ello sea índice de malignidad). El cartílago tiende además a invadir el esponjoso de la exostosis, siendo más blando, jugoso, grisáceo y transparente que el cartílago hialino normal. En fases más avanzadas, se ve una gran masa abollonada, en coliflor, bien delimitadas por unas pseudocápsula fibrosarcoma que tiende a envolver la base de la exostosis y el hueso de implantación. El tumor está más intensamente calcificado en profundidad en las zonas en que las calcificaciones aparecen con su aspecto habitual blanco amarillento, gredoso y granuloso, de consistencia dura; la osificación del tumor tiene el aspecto de un hueso esponjoso hiperémico o ebúrneo y blanquecino. Cuando nuestra calcificado (en las zonas superficiales del tumor y en los tumores de crecimiento más activo y de mayor grado de malignidad), el cartílago aparece como un conjunto de lóbulos voluminosos e irregulares, de aspecto jugoso, tan lúcido, a veces con un reblandecimiento mucoide hacía el centro lobular.
Histológicamente casi siempre está constituido por cartílago bien diferenciado. Excepcionalmente se pueden observar zonas mixoides. Lo mismo que el condrosarcoma central también presenta 3 grados y, si bien el grado III es excepcional y se observa casi exclusivamente cuando hay progresión de la malignidad en las recidivas.
El diagnóstico histológico siempre debe tener en cuenta los datos clínicos, radiológicos, gammagráficos y macroscópicos. El TAC y la RM aporta elementos muy útiles para el diagnóstico diferencial entre exostosis y condrosarcoma periférico. Puede haber dudas con la condromatosis sinovial agresiva cuyo aspecto macroscópico puede ser igual al de un condrosarcoma y la citología puede presentar aspectos que harían pensar en un grado 1 e incluso en un grado 2. La diferencia reside en la localización articular de la condromatosis y en que su arquitectura histológica es distinta. Este diagnóstico diferencial es muy importante, ya que el tratamiento de la condromatosis sinovial, aún en su variedad más agresiva, deber ser lo más conservador posible.
También puede darse el caso de tener que hacer diagnóstico diferencial con un osteosarcoma yuxtacortical. En este último, la opacidad a los rayos X. no es la de un cartílago calcificado, sino una a opacidad ósea, más regular y compacta. Además el tronco de implantación sobre el hueso es diferente.
En cuanto a la evolución el crecimiento es bastante lento, llegando en muchas ocasiones, a que la duración de los síntomas hasta la intervención es mayor de dos años. La recidiva local puede manifestarse hasta incluso los diez años. La muerte puede producirse al cabo de veinte o treinta años después de los primeros síntomas. Las metástasis pulmonares son poco frecuentes (menos del 20% de los casos) y en general tardías. Las metástasis en los ganglios regionales son rarísimas. Hay casos en los que se han manifestado en el transcurso de los años numerosas recidivas, sin ninguna metástasis estas recidivas también pueden curarse con intervención quirúrgica amplia.
Para su tratamiento requieren una intervención amplia. En ocasiones esta no es realizable por adherencias de un tumor a la red vascular y nerviosa, a la pared de la vejiga o al peritoneal. En estos casos se realiza una resección parcial con riesgo elevado de recidiva. En las localizaciones en la columna vertebral, es muy difícil poder realizar algo más que una intervención marginal.
El pronóstico depende de la posibilidad de exéresis amplia y del grado histológico. Por tanto excepto el grado 3 que produce metástasis, y por otro lado que es muy raro, el pronóstico de los condrosarcoma periféricos localizado en los miembros es bueno, ya que es posible la resección amplia y eventualmente la amputación.
Condrosarcoma Mesenquimal:
Es menos como un que condrosarcoma de células claras. Normalmente la mayoría nace en el hueso pero también se han y informado en los tejidos blandos. Afecta a los huesos  craneofaciales (mandíbula), pelvis, costillas, columna y fémur. Las el porcentaje por sexo y edad muy al no difiere de los otros condrosarcomas. Radiológicamente es osteolítico y de forma ocasional puede presentar calcificaciones intratumorales. Histológicamente está compuesto de nódulos de cartílago de apariencia benigna dentro de un fondo de células redondas pequeñas indiferenciadas. El componente indiferenciado de células redondas pequeñas tiene a menudo la apariencia histológica de un hemangiopericitoma. En una revisión de 111 casos los pacientes con condrosarcoma mesenquimal, Nakashima et al informaron que la proporción de supervivencia a los cinco años después de una resección amplía era del 60% y la supervivencia a los diez años era del 25%. En este grupo de pacientes 57 eran hembras y la edad oscilaba entre 4 y 64 años, la localización ósea más común era las costillas, columna, pelvis y fémur. El condrosarcoma mesenquimal se trata con una resección quirúrgica.
craneofaciales (mandíbula), pelvis, costillas, columna y fémur. Las el porcentaje por sexo y edad muy al no difiere de los otros condrosarcomas. Radiológicamente es osteolítico y de forma ocasional puede presentar calcificaciones intratumorales. Histológicamente está compuesto de nódulos de cartílago de apariencia benigna dentro de un fondo de células redondas pequeñas indiferenciadas. El componente indiferenciado de células redondas pequeñas tiene a menudo la apariencia histológica de un hemangiopericitoma. En una revisión de 111 casos los pacientes con condrosarcoma mesenquimal, Nakashima et al informaron que la proporción de supervivencia a los cinco años después de una resección amplía era del 60% y la supervivencia a los diez años era del 25%. En este grupo de pacientes 57 eran hembras y la edad oscilaba entre 4 y 64 años, la localización ósea más común era las costillas, columna, pelvis y fémur. El condrosarcoma mesenquimal se trata con una resección quirúrgica.
Condrosarcoma Desdiferenciado
Es un tipo raro de condrosarcoma de alto grado que se origina de un condrosarcoma de bajo grado recidivante (central o periférico) o de una lesión benigna del cartílago.  Representa aproximadamente el 15% de condrosarcomas centrales y el 4% de condrosarcomas periféricos.
Representa aproximadamente el 15% de condrosarcomas centrales y el 4% de condrosarcomas periféricos.
Clínicamente se comporta como un tumor maligno de alto grado con fracturas patológicas dolor y tumefacción por extensión a los tejidos blandos. Estas manifestaciones pueden haber estado precedidas por las de un tumor benigno o maligno de bajo grado. Se da en personas mayores de 50 años.
En el examen histológico tiene áreas de células fusiformes malignas que no pueden ser reconocidas como de origen cartilaginoso, adyacente a las áreas de condrocitos neoplásicos rodeado por una matriz de cartílago hialino. Un sarcoma de alto grado de células fusiformes coexiste con un tumor condroide de bajo grado; histológicamente habrá áreas con histiocitoma fibroso maligno, osteosarcoma, o fibrosarcoma. Para el diagnóstico anatomopatológico preciso hará falta el estudio de la totalidad del tumor.
El condrosarcoma desdiferenciado, que es el más malignos de los condrosarcoma, tiene un riesgo muy elevado de provocar metástasis a distancia. En una revisión de la Clínica Mayo encontraron que sólo 8 de 68 pacientes sobrevivió más de cinco años después del diagnóstico inicial. Sólo 5% de pacientes sobrevivirán más de 5 años.
El tratamiento debe ser lo más agresivo posible mediante una intervención quirúrgica amplia o incluso radical.
Su origen. Si es primario o secundario.
Los condrosarcomas primarios aparecen de novo y no se asocian a una lesión preexistente, mientras que los secundarios se levantan televisiones preexistentes benignas del cartílago que pueden ser un encondroma, una exostosis osteocartilaginosa un fibroma condromixoide, una condromatosis sinovial, un condromas periostal, o incluso un condroblastoma. Sólo los pacientes con exostosis osteocartilaginosa hereditarias múltiples o encondromas múltiples (enfermedad de Ollier) tienen un riesgo reconocido de desarrollar un condrosarcoma secundario. Schwartz et al. informo que la proporción de pacientes con enfermedad Ollier que desarrollará un condrosarcoma es del 25% en la edad de 40 años. Los pacientes con enfermedad de Maffucci (encondromas  múltiples y hemangiomas) tienen 100% del riesgo de desarrollar un tumor maligno (sarcoma o carcinoma) durante su vida. Los condrosarcomas secundarios que se levanta de una exostosis osteocartilaginosa tienen mejor pronóstico que otros condrosarcoma y raramente dan metástasis; de hecho, las únicas metástasis de condrosarcoma que surge de una exostosis osteocartilaginosa solitaria han sido aquellas aquellos con un grado histológico alto o un componente desdiferenciado. Una exostosis osteocartilaginosa que aumenta de tamaño en una adulto debe ser resecada, no debido a su potencial metastático, sino a su efecto local y a su riesgo pequeño de desarrollar un condrosarcoma desdiferenciado. La resección debe ser completa y la cubierta cartilaginosa no debe violar ser durante la resección ya que esto puede aumentar el riesgo local de recurrencia.
múltiples y hemangiomas) tienen 100% del riesgo de desarrollar un tumor maligno (sarcoma o carcinoma) durante su vida. Los condrosarcomas secundarios que se levanta de una exostosis osteocartilaginosa tienen mejor pronóstico que otros condrosarcoma y raramente dan metástasis; de hecho, las únicas metástasis de condrosarcoma que surge de una exostosis osteocartilaginosa solitaria han sido aquellas aquellos con un grado histológico alto o un componente desdiferenciado. Una exostosis osteocartilaginosa que aumenta de tamaño en una adulto debe ser resecada, no debido a su potencial metastático, sino a su efecto local y a su riesgo pequeño de desarrollar un condrosarcoma desdiferenciado. La resección debe ser completa y la cubierta cartilaginosa no debe violar ser durante la resección ya que esto puede aumentar el riesgo local de recurrencia.

- Su localización en el hueso: central o periférico.
Los condrosarcomas centrales aparecen desde del medular y los periféricos surgen de la superficie del hueso. Para los propósitos prácticos, sólo los condrosarcomas secundarios necesitan ser subdivididos en central periférico; los condrosarcomas primarios casi siempre son centrales. La mayoría de los condrosarcomas periféricos son secundarios a una exostosis osteocartilaginosa, y los condrosarcomas centrales secundarios casi siempre derivan de un encondroma el tratamiento y pronóstico de los condrosarcomas secundarios centrales son idénticas a las del condrosarcoma primario.
Condrosarcoma Periférico
Se trata de un condrosarcoma secundarios que nace de una exostosis, solitaria o múltiple con predominio en el sexo masculino y que siempre será después de la pubertad.
Tienen un buen pronóstico, mejor que el de otros condrosarcomas y raramente da metástasis
Se localiza preferentemente en la pelvis, fémur proximal, columna vertebral y sacro, húmero proximal, costillas, omóplato y fémur distal. Es raro observarlo en la rodilla, por otro lado lugar frecuente de exostosis. En la pelvis se origina más frecuentemente en el ala y días como en el marco anterior. En las vértebras y desacuerdo en el arco posterior. En los huesos largos, se origina siempre en una extremidad diafisarias próxima a la metáfisis. El tumor se extiende por el exterior del hueso, invaden la en exostosis preexistente y, en las formas más agresivas y más avanzadas, también del hueso de implantación.
El signo más importante es la presencia de una masa adherida al hueso y de consistencia y dura, abollonada, de contornos nítidos y que se desarrolla con lentitud. En algún caso el enfermo sufre una exostosis múltiple, en otros casos, estaría perfecto de una exostosis solitario, pero lo más habitual es no encontrar en la anamnesis ningún rastro de exostosis preexistente. La masa que es indolora y en algún caso puede alcanzar dimensiones gigantescas.
Radiográficamente la imagen típica está formada por núcleos opacos debidos a calcificaciones y osificaciones del tejido neoplásico. Las calcificaciones representan en forma de gránulos, anillos o de ” palomitas de maíz”. Las osificaciones están más que estructuradas y forman “sarmientos” culpa indios, imágenes de tabiques en “migas de pan” grumos de opacidad intensa. Se presenta por lo tanto una masa extraóseo única lobulada, abollonada en “coliflor” e intensamente y opaca a los rayos X.. La capa superficial del tumor, de formación más reciente, están menos calcificado y, por consiguiente, puede presentar contornos vagos hacia las partes blandas. Con la ayuda del TAC y la RM puede observarse la parte cartilaginosa no calcificada del tumor. Esta parte cartilaginosa puede representar la mayor parte de la neoplasia. Estas técnicas de diagnóstico puede asimismo demostrara que se trata del primer estadio del paso de la exostosis han condrosarcoma periférico, haciendo visible un aumento del espesor del capuchón cartilaginosa de una exostosis que en la radiografía parece más o menos típica. Asimismo estas técnicas son las únicas que pueden permitir detectar la presencia de nódulos cartilaginosos de recidiva local de tumor en las partes blandas tras una exéresis quirúrgica y inadecuada.
 El condrosarcoma periférico muestra una importante hipercaptación en la gammagrafía, generalmente más que la de la exostosis. Cuando el condrosarcoma periférico en vuelve al hueso de origen, habiendo invadido y borrado completamente la exostosis, el tronco de implantación es irreconocible y se ve como, en ocasiones, el tumor y invade el tejido esponjoso de hueso contiguo.
El condrosarcoma periférico muestra una importante hipercaptación en la gammagrafía, generalmente más que la de la exostosis. Cuando el condrosarcoma periférico en vuelve al hueso de origen, habiendo invadido y borrado completamente la exostosis, el tronco de implantación es irreconocible y se ve como, en ocasiones, el tumor y invade el tejido esponjoso de hueso contiguo.
Macroscópicamente, cuando en un adulto, la envoltura cartilaginosa de una exostosis alcanza o sobrepasa 1 o 2 cm de espesor, hay que sospechar una malignización (en el niño, el espesor puede alcanzar los 2 cm sin que por ello sea índice de malignidad). El cartílago tiende además a invadir el esponjoso de la exostosis, siendo más blando, jugoso, grisáceo y transparente que el cartílago hialino normal.
En fases más avanzadas, se ve una gran masa abollonada, en coliflor, bien delimitadas por unas pseudocápsula fibrosarcoma que tiende a envolver la base de la exostosis y el hueso de implantación. El tumor está más intensamente calcificado en profundidad en las zonas en que las calcificaciones aparecen con su aspecto habitual blanco amarillento, gredoso y granuloso, de consistencia dura; la osificación del tumor tiene el aspecto de un hueso esponjoso hiperémico o ebúrneo y blanquecino. Cuando nuestra calcificado (en las zonas superficiales del tumor y en los tumores de crecimiento más activo y de mayor grado de malignidad), el cartílago aparece como un conjunto de lóbulos voluminosos e irregulares, de aspecto jugoso, tan lúcido, a veces con un reblandecimiento mucoide hacía el centro lobular.

Histológicamente casi siempre está constituido por cartílago bien diferenciado. Excepcionalmente se pueden observar zonas mixoides. Lo mismo que el condrosarcoma central también presenta 3 grados y, si bien el grado III es excepcional y se observa casi exclusivamente cuando hay progresión de la malignidad en las recidivas.
El diagnóstico histológico siempre debe tener en cuenta los datos clínicos, radiológicos, gammagráficos y macroscópicos. El TAC y la RM aporta elementos muy útiles para el diagnóstico diferencial entre exostosis y condrosarcoma periférico. Puede haber dudas con la condromatosis sinovial agresiva cuyo aspecto macroscópico puede ser igual al de un condrosarcoma y la citología puede presentar aspectos que harían pensar en un grado 1 e incluso en un grado 2. La diferencia reside en la localización articular de la condromatosis y en que su arquitectura histológica es distinta. Este diagnóstico diferencial es muy importante, ya que el tratamiento de la condromatosis sinovial, aún en su variedad más agresiva, deber ser lo más conservador posible.
También puede darse el caso de tener que hacer diagnóstico diferencial con un osteosarcoma yuxtacortical. En este último, la opacidad a los rayos X. no es la de un cartílago calcificado, sino una a opacidad ósea, más regular y compacta. Además el tronco de implantación sobre el hueso es diferente.
En cuanto a la evolución el crecimiento es bastante lento, llegando en muchas ocasiones, a que la duración de los síntomas hasta la intervención es mayor de dos años. La recidiva local puede manifestarse hasta incluso los diez años. La muerte puede producirse al cabo de veinte o treinta años después de los primeros síntomas. Las metástasis pulmonares son poco frecuentes (menos del 20% de los casos) y en general tardías. Las metástasis en los ganglios regionales son rarísimas. Hay casos en los que se han manifestado en el transcurso de los años numerosas recidivas, sin ninguna metástasis estas recidivas también pueden curarse con intervención quirúrgica amplia.
Para su tratamiento requieren una intervención amplia. En ocasiones esta no es realizable por adherencias de un tumor a la red vascular y nerviosa, a la pared de la vejiga o al peritoneal. En estos casos se realiza una resección parcial con riesgo elevado de recidiva. En las localizaciones en la columna vertebral, es muy difícil poder realizar algo más que una intervención marginal.
 El pronóstico depende de la posibilidad de exéresis amplia y del grado histológico. Por tanto excepto el grado 3 que produce metástasis, y por otro lado que es muy raro, el pronóstico de los condrosarcoma periféricos localizado en los miembros es bueno, ya que es posible la resección amplia.
El pronóstico depende de la posibilidad de exéresis amplia y del grado histológico. Por tanto excepto el grado 3 que produce metástasis, y por otro lado que es muy raro, el pronóstico de los condrosarcoma periféricos localizado en los miembros es bueno, ya que es posible la resección amplia.
