El Sarcoma Pleomórfico Indiferenciado (UPS), antiguamente fibrohistiocitoma maligno (FHM), es un cáncer de origen incierto que aparece tanto en las partes blandas como en el hueso. Fue descrito por vez primera por Kauffman y Stout en 1962 y desde ese momento ha sido objeto de controversia. Estos autores describieron el FHM como un tumor rico en histiocitos con patrón de crecimiento estoriforme. Ya en 1977 el FHM era considerado el sarcoma de partes blandas más frecuente en los adultos. Pese a su frecuente diagnóstico, el FHM continúa siendo un enigma. No ha conseguido identificarse aún la verdadera célula que da origen de este tumor. En 2002, la Organización Mundial de la Salud (OMS) desclasificó el FHM como entidad con categoría diagnóstica y pasó a denominarlo sarcoma pleomórfico indiferenciado, no especificado, NOS.4
Esta nueva terminología ha venido sustentada por un compendio de evidencias acumuladas a lo largo de la última década que sugieren que el FHM constituye la ruta final común de distintos tumores que experimentan una evolución progresiva hacia la desdiferenciación. 5,12,15,19

Figura 2
Figura 2: Un ejemplo histológico de la variante mixoide del FHM…
El FHM muestra un amplio rango de apariencias histológicas, existiendo cuatro subtipos reconocidos descritos:3
– Pleomórfico-estoriforme
– Mixoide
– De células gigantes
– Inflamatorio
De entre ellos, el tipo pleomórfico-estoriforme es el más frecuente, constituyendo hasta un 70% de los casos (Figura 1). La variante mixoide es la segunda en frecuencia, constituyendo aproximadamente un 20% de los casos (Figura 2).
A diferencia de otros subtipos de FHM, la variante mixoide tiende a ser menos agresiva y por consiguiente se asocia a un mejor pronóstico. Las variantes inflamatoria y de células gigantes son muy poco frecuentes. El fibrohistiocitoma maligno inflamatorio suele aparecer en el retroperitoneo.
La variante mixoide debe, por definición, contener al menos un 50% de áreas mixoides. En algunos casos un nódulo tumoral puede tener aspecto mixoide en toda su extensión, lo que causa confusión diagnóstica con otras entidades menos agresivas como el mixoma o la fascitis nodular. La mayoría de los tumores incluidos en la variante mixoide del FHM se comportan como neoplasias de bajo grado y por tanto muestran un curso clínico menos agresivo.
¿Cómo se Presenta el Sarcoma Pleomórfico Indiferenciado?
Como ocurre con todos los sarcomas de hueso y de partes blandas, el fibrohistiocitoma maligno es raro, diagnosticándose cada año sólo unos pocos miles de casos.
El fibrohistiocitoma maligno de partes blandas se presenta de forma típica en un paciente de entre 50 y 70 años, aunque puede aparecer a cualquier edad, es muy raro en personas de menos de 20 años de edad.
Hay un ligero predominio del sexo masculino. El fibrohistiocitoma maligno de partes blandas puede aparecer en cualquier parte del organismo, pero la localización más habitual es la extremidad inferior, especialmente el muslo. Otras localizaciones frecuentes son la extremidad superior y el retroperitoneo. Los pacientes suelen referir la aparición en un breve periodo de tiempo (que oscila entre varias semanas y algunos meses) de una tumefacción o masa. No es infrecuente que los pacientes describan un traumatismo previo en la zona afectada. Por ejemplo, los pacientes relatarán que “se golpearon con la esquina de una mesa” y que desde entonces notan un bulto en muslo. Hasta donde sabemos los traumatismos no son la causa del FHM, pero dirigen la atención hacia la extremidad afectada. La masa no produce generalmente ningún dolor salvo que comprima un nervio cercano. Otros síntomas, como la fatiga y la pérdida de peso, no son habituales, pero pueden aparecer en pacientes con enfermedad avanzada. Los tumores de localización retroperitoneal pueden alcanzar un tamaño muy grande antes de ser detectados y los pacientes no sienten una masa como tal, sino síntomas constitucionales asociados al tumor, como anorexia o aumento de la presión abdominal.
Dado que el fibrohistiocitoma maligno es extremadamente raro en niños, antes de asumir este diagnóstico en la población pediátrica deberán agotarse otras posibilidades.

Figura 3
Figura 3: Aspecto clínico de un FHM de la región proximal lateral del muslo…
“Noto un Bulto. ¿Y Ahora, Qué?”
No todos los bultos o masas son finalmente cancerosos y, en realidad, la mayoría no lo son. No obstante, todas las tumoraciones deben ser motivo de consulta médica. Su médico habrá de hacerle una serie de preguntas relacionadas con la aparición de la masa, tales como “¿cuánto tiempo hace que nota usted el bulto?” y “¿percibe usted que está aumentando de tamaño?”. “Si es así, ¿desde hace cuánto tiempo?” El o ella llevarán entonces a cabo un exhaustivo examen físico que valorará el tamaño y la consistencia de la tumoración, explorando además otras zonas de la extremidad afectada y buscando posibles ganglios linfáticos aumentados de tamaño.

Figura 4
Figura 4: Imagen clínica de un FHM de la mano…
Con frecuencia, la primera prueba de imagen realizada es una radiografía. Habitualmente, ésta se sigue de una resonancia magnética (RM). La RM es la técnica más útil para visualizar tumores de partes blandas y proporciona una información sumamente valiosa sobre la masa, como su tamaño, localización y proximidad a estructuras neurovasculares. Es importante señalar que no puede diagnosticarse un tumor maligno únicamente a partir de la resonancia. Para aquellos pacientes en los que no puede realizarse una resonancia magnética (por tener implantes metálicos, como un marcapasos) puede recurrirse al TAC.
Tras analizar toda la información obtenida, y si la masa continua siendo sospechosa de ser un sarcoma, el paciente deberá ser remitido a un especialista en este tipo de neoplasia. Éste, que habitualmente será un traumatólogo o un cirujano general, realizará pruebas adicionales y planificará la obtención de una biopsia.
 Figura 5
Figura 5
Figura 5a (izquierda): Proyección axial mediante RMN de un FHM del muslo…
Cuando se llega a un diagnóstico de sarcoma, es importante determinar si la neoplasia es un tumor aislado (localizado) o diseminado (metastásico). Cuando un sarcoma de partes blandas se disemina, habitualmente metastatiza en el pulmón. Por consiguiente, de forma rutinaria se realiza un TAC de tórax para determinar la presencia o ausencia de metástasis. Aunque los sarcomas, incluyendo el FHM, pueden extenderse a otras localizaciones como los ganglios linfáticos o el hueso, este hecho es bastante infrecuente. El papel desempeñado por pruebas como la gammagrafía ósea o el PET-TAC no está aún completamente definido.20,23,30 Hay una considerable variabilidad acerca de la utilidad clínica que los médicos conceden a este tipo de técnicas adicionales.
La inmensa mayoría de los casos de diseminación metastásica en los sarcomas (incluyendo el FHM) se presenta como enfermedad pulmonar (90%). Las metástasis en localizaciones extrapulmonares son infrecuentes, pudiendo ocurrir en los ganglios linfáticos (10%), hueso (8%) e hígado (1%).
La técnica de PET-TAC se fundamenta en la conocida alta actividad metabólica de las células neoplásicas. Este procedimiento emplea un análogo de la glucosa marcado radiactivamente, denominado fluoro-deoxi-D-glucosa (FDG).18 que es metabolizado en tasas más altas por las células tumorales. El consumo del FDG se expresa como valor máximo estándar de captación (SUV). Aunque es un procedimiento con una alta sensibilidad, no es específico para el diagnóstico de los sarcomas. Varios estudios de investigación han tratado de definir el papel del PET-TAC en los sarcomas de partes blandas.13,33,34 A medida que vaya desarrollándose nuestro conocimiento y comprensión de esta nueva técnica, irá definiéndose con más precisión su capacidad diagnóstica y de estadificación en este tipo de neoplasias. Por el momento, el balance coste-efectividad y el papel definitivo del PET-TAC están aún por definir con claridad.
 Figura 6
Figura 6
Figura 6: Biopsia con aguja de un FHM del muslo…
La Biopsia del Fibrohistiocitoma Maligno
La biopsia es el procedimiento mediante el cual se obtiene el tejido necesario para obtener un diagnóstico. Las biopsias pueden ser realizadas mediante distintos procedimientos. La biopsia con aguja consiste en la inserción en el tumor de una aguja de pequeño calibre para obtener una muestra de éste.17,45 Las biopsias con aguja pueden ser realizadas habitualmente en la consulta clínica (ver figura 6). Si el tumor asienta en una localización en la que es difícil de palpar o hay en la proximidad del tumor estructuras que pudieran ser dañadas, se procederá a realizar la biopsia con aguja bajo control radiológico guiado con TAC.
La biopsia abierta es un procedimiento quirúrgico que normalmente se realiza en el quirófano bajo sedación o anestesia. En las biopsias incisionales se extrae solo una pequeña parte del tumor para ser analizada. En las biopsias escisionales se extirpa la totalidad del tumor. Las biopsias escisionales se reservan habitualmente para tumores pequeños (menores de 3 cm).
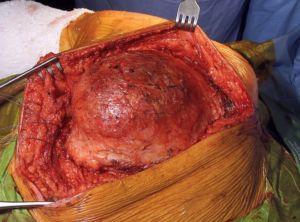 Figura 7
Figura 7
Figura 7: Incisión de conservación de la extremidad en el miembro superior…
La decisión de extirpar parte de la masa o su totalidad en el momento de la biopsia es de trascendencia crítica. Nunca se insistirá lo suficiente sobre la importancia de disponer de un especialista en sarcomas tanto para llevar a cabo la toma de la biopsia como para orientar al cirujano en la planificación de la misma.
La biopsia es con frecuencia el primer paso de un procedimiento exitoso de conservación de la extremidad. La localización de la incisión para realizar la biopsia y los aspectos técnicos relacionados con la obtención del tejido necesario para el diagnóstico pueden tener un gran impacto en las intervenciones subsiguientes.28
El tejido obtenido en la biopsia es evaluado por un patólogo. El patólogo emplea diversas herramientas y técnicas diagnósticas que incluyen la microscopía óptica, la inmunohistoquímica, el microscopio electrónico y los estudios moleculares como procedimientos para emitir un diagnóstico. Además de hacer un diagnóstico histológico, el patólogo suministra un dato fundamental: el grado del tumor. El grado tumoral hace referencia a la apariencia microscópica de la neoplasia y es un reflejo de la agresividad de ésta. Los tumores de alto grado se comportan de manera más agresiva, lo que significa que tienen una tendencia más elevada a recidivar (o “reproducirse”) y a metastatizar. Los tumores de bajo grado son menos agresivos y tienen menor tendencia a recidivar y a diseminarse. El grado no es una garantía absoluta del comportamiento del tumor pero constituye sin duda uno de los factores que ayudan al equipo de tratamiento a elaborar sus recomendaciones.
Se han descrito varias anomalías citogenéticas en los sarcomas de partes blandas incluyendo el FHM. Más del 80% de los FHM presentan mutaciones que afectan a los locus cromosómicos 1q31, 9q31, 5p14 y 7q32. Se han descrito también anomalías en 3p12, 11p11 y 19p13. En el análisis multivariante según tamaño y grado tumoral, la ganancia de 7q32 parece predecir una peor supervivencia.24
Estadificación
Una vez que se han realizado todas las técnicas de imagen y la toma biópsica, podrá determinarse el estadio de la enfermedad. El sistema de estadificación más común es el de la AJCC (American Joint Commitee on Cancer) para sarcomas de partes blandas (ver tabla 1).1 Normalmente, el paciente preguntará sobre el estadio de su enfermedad. Deberá tenerse presente que el estadio no es una garantía del potencial comportamiento del tumor; la estadificación únicamente proporciona al equipo de tratamiento una guía para el mejor manejo del paciente destinada a optimizar el pronóstico clínico.
Una vez que se ha confirmado el diagnóstico de FHM se elabora un plan de tratamiento individualizado para cada paciente. El tratamiento del sarcoma requiere un abordaje multidisciplinario y por consiguiente será necesaria la participación de un equipo médico completo en el cuidado del paciente. Existen básicamente tres formas principales de tratamiento que necesitarán ser coordinadas para tratar el UPS:
Cirugía
Radioterapia
Quimioterapia
Cirugía
La cirugía es la piedra angular del tratamiento de todos los sarcomas de partes blandas. El objetivo de la cirugía es eliminar toda la enfermedad del área afectada. Para los sarcomas de las extremidades, las alternativas quirúrgicas se centran en dos categorías: conservación de la extremidad y amputación. En el pasado los sarcomas de partes blandas eran tratados mediante la amputación. Sin embargo, de la misma manera que nuestros conocimientos sobre el sarcoma han ido evolucionando lo ha hecho su tratamiento. Varios estudios han demostrado que no hay diferencias en la supervivencia de los pacientes tratados con amputación frente a aquéllos en los que se realiza cirugía conservadora de la extremidad.46-47 En un ensayo clínico aleatorio llevado a cabo por el Instituto nacional del Cáncer40 no se encontraron diferencias significativas en la supervivencia global de los pacientes con sarcomas de partes blandas que sufrieron amputación frente a aquellos a los que no se les realizó (71%). En la actualidad casi el 90% de los tumores son extirpados mediante cirugía conservadora de la extremidad, lo que implica que el tumor se extrae salvando al mismo tiempo el miembro afectado. La cirugía conservadora de la extremidad deberá realizarse únicamente si el cirujano tiene la certeza de que el tumor puede ser extirpado en su totalidad y que la extremidad remanente tendrá una capacidad funcional aceptable. Obviamente, conciliar el pronóstico oncológico y funcional es un procedimiento complejo y subjetivo, difícil de asumir. Es muy importante que el paciente y el cirujano que vaya a intervenirle discutan de forma previa a la operación las expectativas reales de todas las opciones quirúrgicas existentes. Dependiendo del tamaño del tumor y de las estructuras que hayan de ser sacrificadas, en ocasiones es necesaria la reconstrucción tras la extirpación quirúrgica del tumor. Por ejemplo, puede necesitarse reconstruir un hueso o una articulación o bien crear injertos de partes blandas para cubrir una herida quirúrgica.
Una vez que el tumor ha sido extirpado, el patólogo analiza la pieza en su totalidad a fin de confirmar el grado histológico y el estado de los márgenes. El término “margen” hace referencia a los bordes externos de la pieza de resección. Un margen negativo significa que no hay células tumorales en la periferia del tumor e implica que se ha efectuado una resección completa. Un margen positivo significa que se han encontrado células tumorales en el borde de la pieza quirúrgica, lo que supone la probable existencia de enfermedad microscópica residual. Como es obvio, se espera obtener márgenes negativos en el momento de la extirpación quirúrgica en todos los casos, pero desafortunadamente no siempre es posible conseguirlo. Los procedimientos quirúrgicos empleados en los sarcomas se clasifican tal y como consta en la tabla 2. Siempre y cuando sea posible se realizarán técnicas quirúrgicas radicales mediante resecciones amplias a fin de conseguir márgenes negativos.
Radioterapia
La radioterapia es administrada por el oncólogo radioterapeuta. El objetivo de la radiación es mejorar el control local del tumor eliminando la enfermedad microscópica residual. La radioterapia ha demostrado con claridad ser capaz de disminuir la incidencia de recidiva local y se ha convertido en parte integrante del tratamiento del FHM.6,27 Las dosis habituales de radiación oscilan entre 45 Gy y 65 Gy.
En un ensayo aleatorio prospectivo del NCI, 91 pacientes con tumores de alto grado fueron distribuidos aleatoriamente en grupos en los que se les trató bien solo con cirugía, bien con cirugía y radioterapia con irradiación externa post-quirúrgica, o solo radioterapia.48 Los pacientes del grupo tratado únicamente con cirugía tuvieron tasas de recidiva local del 20%, frente al 0% de recidivas aparecidas en el grupo que se trató con cirugía más radioterapia. No se observaron diferencias en la supervivencia global entre ambos grupos. En términos generales, en los pacientes tratados con una adecuada cirugía de conservación de la extremidad complementada con radioterapia, la probabilidad de conseguir el control local de la enfermedad es del 85%.

Figura 8
Figura 8: Imagen intraoperatoria de un FHM de la escápula…
Hay diferentes formas de administrar la radioterapia. El procedimiento más común es la radioterapia mediante irradiación externa, que puede ser suministrada de forma preoperatoria, intraoperatoria o postoperatoria, o de manera combinada. Cada una de ellas tiene ventajas e inconvenientes (ver tabla 3). En aquellos tumores que están en contacto con nervios principales y/o vasos sanguíneos la radioterapia preoperatoria puede potencialmente “encoger” el tumor, haciendo posible o más sencilla la cirugía con preservación de la extremidad. El principal inconveniente de la radioterapia preoperatoria es su asociación con complicaciones postoperatorias de la herida quirúrgica.10,35 La radioterapia postoperatoria es, probablemente, la modalidad empleada con más frecuencia. De forma clásica, la radioterapia pre y postoperatoria son administradas durante periodos de 5 semanas. La radioterapia intraoperatoria tiene la ventaja de ser capaz de liberar una alta dosis de radiación directamente sobre el área concreta afectada, respetando al mismo tiempo órganos adyacentes como el intestino o la vejiga. Esto es especialmente útil en el tratamiento de grandes sarcomas retroperitoneales en los que es sumamente difícil conseguir el control local de la enfermedad.43
Otro modo de suministrar la radiación terapéutica es mediante una técnica denominada braquiterapia. Una vez que el cirujano ha extirpado el tumor, el oncólogo radioterapeuta coloca tubos vacíos en el lecho quirúrgico. Cuando la herida quirúrgica comienza a cicatrizar (aproximadamente 5 días después de la cirugía), los catéteres se rellenan con materiales radioactivas que quedan depositados en el lecho quirúrgico durante 5 días. Ello permite el suministro de altas dosis de radiación en un corto periodo de tiempo, evitando la necesidad de desplazarse diariamente durante varias semanas para recibir el tratamiento radioterápico.
En un ensayo aleatorio prospectivo del NCI, 91 pacientes con tumores de alto grado fueron distribuidos aleatoriamente en grupos en los que se les trató bien sólo con cirugía bien con cirugía y radioterapia con irradiación externa post-quirúrgica, o solo radioterapia.48 Los pacientes del grupo tratado únicamente con cirugía tuvieron tasas de recidiva local del 20%, frente al 0% de recidivas aparecidas en el grupo que se trató con cirugía más radioterapia. No se observaron diferencias en la supervivencia global entre ambos grupos. En términos generales, en los pacientes tratados con una adecuada cirugía de conservación de la extremidad complementada con radioterapia, la probabilidad de conseguir el control local de la enfermedad es del 85%.
Desafortunadamente la radioterapia tiene efectos secundarios bien conocidos. Los problemas relacionados con la cicatrización de la herida se han descrito ya previamente. Otras complicaciones como la cicatrización exagerada del tejido, que causa de forma secundaria rigidez y dureza muscular, así como la decoloración de la piel, han sido también descritas. La complicación más grave derivada de la radioterapia es sin embargo la aparición de una neoplasia secundaria en el campo de radiación.8,29,32 Estas neoplasias se denominan sarcomas post-radiación o sarcomas radioinducidos. Los sarcomas radioinducidos son poco frecuentes y aparecen en menos del 5% de los pacientes que sobreviven a largo plazo.
Quimioterapia
El papel de la quimioterapia en el tratamiento del FMH no está aún claramente definido. Varios ensayos clínicos incorporando un fármaco quimioterápico denominado adriamicina (conocido también como doxorrubicina) han demostrado una tendencia a la mejoría en la supervivencia libre de enfermedad sin un impacto relevante asociado sobre la supervivencia global. Los resultados de un gran meta-análisis que incluyó casi 1600 pacientes con sarcoma de partes blandas concluyó que la adición de quimioterapia mejoraba la supervivencia global en menos del 10%.2 Los resultados fueron mejores en pacientes con tumores localizados en las extremidades que en aquéllos con neoplasias localizadas en el retroperitoneo o en el tronco. Más recientemente algunos ensayos clínicos que incorporaban ifosfamida y doxorrubicina han demostrado una mejoría en la supervivencia libre de enfermedad.26,36 Una de las mayores limitaciones de la quimioterapia es la relacionada con los efectos secundarios que aparecen tras el suministro de las dosis necesarias para obtener un impacto significativo en la supervivencia global específica de enfermedad. La inclusión de fármacos de soporte como los factores de crecimiento hemoyéticos ha permitido suministrar dosis más altas de quimioterápicos, observándose una tendencia a la mejoría en la supervivencia.
Desafortunadamente la interpretación de éstos y otros resultados de los ensayos con quimioterápicos ha variado tanto que se ha hecho muy dificultoso para los pacientes descifrar la información con el fin de tomar decisiones en relación a la inclusión de la quimioterapia como parte de su tratamiento. En cualquier caso, la decisión de incorporar la quimioterapia al tratamiento del FHM deberá hacerse siempre bajo la supervision de un oncólogo médico. La quimioterapia debería probablemente suministrarse a aquellos pacientes que han padecido ya enfermedad metastásica o que tienen un riesgo máximo de desarrollar metástasis. Con mayor probabilidad, la quimioterapia será suministrada en el contexto de un ensayo clínico.
Pronóstico y Evolución
Es bien conocido que los factores pronósticos que se correlacionan con la supervivencia en los pacientes con FHM incluyen el grado tumoral, la profundidad de localización, el tamaño, la existencia o no de metástasis, la edad del paciente y el subtipo histológico.11,16,39 Los factores pronósticos favorables incluyen la edad inferior a 60 años, el tamaño tumoral menor de 5 cm, la localización superficial de la neoplasia, un bajo grado histológico, ausencia de enfermedad metastásica y el subtipo histológico mixoide. Los pacientes mayores con tumores de tamaño superior a 5 cm, de localización profunda y de alto grado histológico no tienen un pronóstico tan favorable. Sin embargo, los pacientes con tumores pequeños de bajo grado conseguirán probablemente la curación completa. En los pacientes con tumores grandes, de localización profunda y de alto grado histológico (estadio III) la supervivencia estimada a los 5 años oscila entre el 34 y el 70%.25,42,49
Nomograma de los Sarcomas
Basándose en un estudio de cohortes prospectivo realizado en pacientes adultos con sarcoma primario de partes blandas (SPB) tratados en el Centro del Cáncer Memorial Sloan-Kettering, se desarrolló un nomograma para predecir la mortalidad específica a los 12 años ligada al sarcoma.21 Las variables del nomograma con valor predictivo incluían la edad al diagnóstico, el tamaño tumoral (menor o igual a 5 cm, de 5 a 10 cm o mayor de 10 cm), el grado histológico (alto o bajo), el subtipo histológico (fibrosarcoma, leiomiosarcoma, liposarcoma, fibrohistiocitoma maligno,tumor maligno de nervio periférico, sarcoma sinovial u otros), la profundidad (superficial o profundo) y la localización (extremidad superior, extremidad inferior, víscera torácica o abdominal, retroperitoneal o cabeza y cuello). La exactitud de este nomograma ha sido validada mediante estándares tanto internos como externos.14 Esta herramienta es susceptible de ser utilizada en pacientes adultos que se encuentran a menos de 6 meses de la cirugía inicial y en los que no hay evidencia de enfermedad metastásica. El nomograma de sarcomas puede ser útil para el asesoramiento a los pacientes, la planificación de su seguimiento y la determinación de criterios de selección de pacientes en los ensayos clínicos.
La recidiva local del tumor (RL) en la misma localización de origen tendrá lugar aproximadamente en un 20-30% del total de pacientes con sarcomas de partes blandas.25,42,49 La recidiva local es menos frecuente en los sarcomas de las extremidades y más probable en los sarcomas retroperitoneales y en los de cabeza y cuello. Esta distribución está directamente relacionada con la posibilidad de extirpar por completo el tumor durante la cirugía. Las tasas más altas de recidiva local se observan en el contexto de casos con márgenes quirúrgicos positivos, que son más difíciles de evitar en localizaciones anatómicas distintas a las extremidades.18 El hecho de si el control local de la enfermedad tiene un impacto definido sobre la supervivencia global no está claro y se mantiene aún en controversia. Se han publicados varios estudios de investigación bien diseñados que apoyan tanto argumentos a favor como en contra de esta cuestión.
Es muy importante para los pacientes comprender que los datos de pronóstico y supervivencia proceden de estudios de análisis retrospectivo enormemente heterogéneos que carecen de criterios de inclusión estrictos. Las estadísticas de supervivencia reflejadas son útiles para el clínico responsable del tratamiento como guía orientativa de la terapia a emplear, pero probablemente tienen valor limitado para un paciente en concreto.
Seguimiento, Tratamiento a Largo Plazo y Supervivencia
Probablemente alrededor de un tercio de los pacientes con FHM de la extremidad y cerca de la mitad de los pacientes con FHM retroperitoneal sufrirán una recidiva, ya sea en su lugar inicial de origen (recidiva local) o en otra localización alejada del primario (recidiva a distancia o metástasis). La mayoría de las recidivas se desarrollan en los primeros 2 años tras el tratamiento, pero pueden ocurrir en cualquier momento a lo largo de la vida del paciente. La frecuencia y duración del seguimiento del paciente varían con arreglo al número de factores de riesgo que tenga de desarrollar una recidiva. Por término medio, el seguimiento de los pacientes es de aproximadamente 10 años. Los pacientes con tumores grandes o de alto grado deberán probablemente ser evaluados inicialmente cada pocos meses, mientras que aquéllos con tumores pequeños o de bajo grado podrán ser revisados de forma anual. Las revisiones de seguimiento incluyen la exploración física y un examen torácico mediante una placa de tórax o un TAC. Dependiendo de las circunstancias puede ser necesaria la realización de una RM del área de origen primario del tumor.
Si se detecta una recidiva, el equipo de tratamiento deberá coordinarse de nuevo para determinar el papel que han de desempeñar la cirugía, la radioterapia y la quimioterapia en el abordaje de ésta. La mayoría de las recidivas locales pueden ser tratadas de forma eficaz con cirugía adicional. Los casos de recidiva local de la enfermedad que puede extirparse quirúrgicamente (aproximadamente, dos tercios del total) disfrutan de una supervivencia a largo plazo prolongada.31,44 Si no se ha administrado previamente radioterapia, el área afectada podrá recibir radiación como tratamiento de la recidiva.
La enfermedad metastásica constituye la forma más grave de recidiva y tiene lugar habitualmente en los pulmones. Los planes individualizados de tratamiento varían de forma significativa dependiendo de un gran número de factores relacionados tanto con el paciente como con su enfermedad, así como de las limitaciones impuestas por tratamientos previos. En los pacientes que tienen metástasis pulmonares aisladas que pueden ser resecadas, es posible conseguir supervivencias prolongadas entre el 20 y el 50% de los casos.7,9,41 La quimioterapia se incorpora con frecuencia en el tratamiento de los pacientes con recidivas a distancia. Desafortunadamente, los pacientes con una recidiva inextirpable tienen mal pronóstico.
BIBLIOGRAFIA
1. In AJCC Cancer Staging Handbook. Edited, New York, Springer-Verlag, 2002.
2. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet, 350(9092): 1647-54, 1997.
3. Enzinger and Weiss’s Soft Tissue Tumors. Edited by Weiss SW, G. J., St. Louis, Mosby, 2001.
4. World Health Organization Classification of Tumors: Pathology and Genetics of Tumors of Soft Tissue and Bone. Edited by Fletcher CDM, U. K., Mertens F., Lyon, France, IARC Press, 2002.
5. Akerman, M.: Malignant fibrous histiocytoma–the commonest soft tissue sarcoma or a nonexistent entity? Acta Orthop Scand Suppl, 273: 41-6, 1997.
6. Barkley, H. T., Jr.; Martin, R. G.; Romsdahl, M. M.; Lindberg, R.; and Zagars, G. K.: Treatment of soft tissue sarcomas by preoperative irradiation and conservative surgical resection. Int J Radiat Oncol Biol Phys, 14(4): 693-9, 1988.
7. Billingsley, K. G.; Burt, M. E.; Jara, E.; Ginsberg, R. J.; Woodruff, J. M.; Leung, D. H.; and Brennan, M. F.: Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg, 229(5): 602-10; discussion 610-2, 1999.
8. Cahan, W. G.; Woodard, H. Q.; Higinbotham, N. L.; Stewart, F. W.; and Coley, B. L.: Sarcoma arising in irradiated bone: report of eleven cases. Cancer, 1(1): 3-29, 1948.
9. Casson, A. G.; Putnam, J. B.; Natarajan, G.; Johnston, D. A.; Mountain, C.; McMurtrey, M.; and Roth, J. A.: Five-year survival after pulmonary metastasectomy for adult soft tissue sarcoma. Cancer, 69(3): 662-8, 1992.
10. Cheng, E. Y.; Dusenbery, K. E.; Winters, M. R.; and Thompson, R. C.: Soft tissue sarcomas: preoperative versus postoperative radiotherapy. J Surg Oncol, 61(2): 90-9, 1996.
11. Coindre, J. M. et al.: Prognostic factors in adult patients with locally controlled soft tissue sarcoma. A study of 546 patients from the French Federation of Cancer Centers Sarcoma Group. J Clin Oncol, 14(3): 869-77, 1996.
12. Dehner, L. P.: Malignant fibrous histiocytoma. Nonspecific morphologic pattern, specific pathologic entity, or both? Arch Pathol Lab Med, 112(3): 236-7, 1988.
13. Eary, J. F.; O’Sullivan, F.; Powitan, Y.; Chandhury, K. R.; Vernon, C.; Bruckner, J. D.; and Conrad, E. U.: Sarcoma tumor FDG uptake measured by PET and patient outcome: a retrospective analysis. Eur J Nucl Med Mol Imaging, 29(9): 1149-54, 2002.
14. Eilber, F. C.; Brennan, M. F.; Eilber, F. R.; Dry, S. M.; Singer, S.; and Kattan, M. W.: Validation of the postoperative nomogram for 12-year sarcoma-specific mortality. Cancer, 101(10): 2270-5, 2004.
15. Fletcher, C. D.: Pleomorphic malignant fibrous histiocytoma: fact or fiction? A critical reappraisal based on 159 tumors diagnosed as pleomorphic sarcoma. Am J Surg Pathol, 16(3): 213-28, 1992.
16. Gaynor, J. J.; Tan, C. C.; Casper, E. S.; Collin, C. F.; Friedrich, C.; Shiu, M.; Hajdu, S. I.; and Brennan, M. F.: Refinement of clinicopathologic staging for localized soft tissue sarcoma of the extremity: a study of 423 adults. J Clin Oncol, 10(8): 1317-29, 1992.
17. Heslin, M. J.; Lewis, J. J.; Woodruff, J. M.; and Brennan, M. F.: Core needle biopsy for diagnosis of extremity soft tissue sarcoma. Ann Surg Oncol, 4(5): 425-31, 1997.
18. Heslin, M. J.; Woodruff, J.; and Brennan, M. F.: Prognostic significance of a positive microscopic margin in high-risk extremity soft tissue sarcoma: implications for management. J Clin Oncol, 14(2): 473-8, 1996.
19. Hollowood, K., and Fletcher, C. D.: Malignant fibrous histiocytoma: morphologic pattern or pathologic entity? Semin Diagn Pathol, 12(3): 210-20, 1995.
20. Jager, P. L.; Hoekstra, H. J.; Leeuw, J.; van Der Graaf, W. T.; de Vries, E. G.; and Piers, D.: Routine bone scintigraphy in primary staging of soft tissue sarcoma; Is it worthwhile? Cancer, 89(8): 1726-31, 2000.
21. Kattan, M. W.; Leung, D. H.; and Brennan, M. F.: Postoperative nomogram for 12-year sarcoma-specific death. J Clin Oncol, 20(3): 791-6, 2002.
22. Kauffman, S. L., and Stout, A. P.: Histiocytic tumors (fibrous xanthoma and histiocytoma) in children. Cancer, 14: 469-82, 1961.
23. Kern, K. A.; Brunetti, A.; Norton, J. A.; Chang, A. E.; Malawer, M.; Lack, E.; Finn, R. D.; Rosenberg, S. A.; and Larson, S. M.: Metabolic imaging of human extremity musculoskeletal tumors by PET. J Nucl Med, 29(2): 181-6, 1988.
24. Larramendy, M. L.; Tarkkanen, M.; Blomqvist, C.; Virolainen, M.; Wiklund, T.; Asko-Seljavaara, S.; Elomaa, I.; and Knuutila, S.: Comparative genomic hybridization of malignant fibrous histiocytoma reveals a novel prognostic marker. Am J Pathol, 151(4): 1153-61, 1997.
25. Le Doussal, V. et al.: Prognostic factors for patients with localized primary malignant fibrous histiocytoma: a multicenter study of 216 patients with multivariate analysis. Cancer, 77(9): 1823-30, 1996.
26. Leyvraz, S.; Bacchi, M.; Cerny, T.; Lissoni, A.; Sessa, C.; Bressoud, A.; and Hermann, R.: Phase I multicenter study of combined high-dose ifosfamide and doxorubicin in the treatment of advanced sarcomas. Swiss Group for Clinical Research (SAKK). Ann Oncol, 9(8): 877-84, 1998.
27. Lindberg, R. D.; Martin, R. G.; Romsdahl, M. M.; and Barkley, H. T., Jr.: Conservative surgery and postoperative radiotherapy in 300 adults with soft-tissue sarcomas. Cancer, 47(10): 2391-7, 1981.
28. Mankin, H. J.; Mankin, C. J.; and Simon, M. A.: The hazards of the biopsy, revisited. Members of the Musculoskeletal Tumor Society. J Bone Joint Surg Am, 78(5): 656-63, 1996.
29. Mark, R. J.; Poen, J.; Tran, L. M.; Fu, Y. S.; Selch, M. T.; and Parker, R. G.: Postirradiation sarcomas. A single-institution study and review of the literature. Cancer, 73(10): 2653-62, 1994.
30. Mekhmandarov, S.; Azaria, M.; Engelberg, S.; and Lieberman, L. M.: Utility of 24 hour bone scan in evaluation of bone involvement by soft tissue sarcoma. Clin Nucl Med, 13(9): 649-51, 1988.
31. Midis, G. P.; Pollock, R. E.; Chen, N. P.; Feig, B. W.; Murphy, A.; Pollack, A.; and Pisters, P. W.: Locally recurrent soft tissue sarcoma of the extremities. Surgery, 123(6): 666-71, 1998.
32. Mills, E. E.: Adjuvant chemotherapy of adult high-grade soft tissue sarcoma. J Surg Oncol, 21(3): 170-5, 1982.
33. Miraldi, F.; Adler, L. P.; and Faulhaber, P.: PET imaging in soft tissue sarcomas. Cancer Treat Res, 91: 51-64, 1997.
34. Nieweg, O. E.; Pruim, J.; van Ginkel, R. J.; Hoekstra, H. J.; Paans, A. M.; Molenaar, W. M.; Koops, H. S.; and Vaalburg, W.: Fluorine-18-fluorodeoxyglucose PET imaging of soft-tissue sarcoma. J Nucl Med, 37(2): 257-61, 1996.
35. O’Sullivan, B. et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet, 359(9325): 2235-41, 2002.
36. Patel, S. R.; Vadhan-Raj, S.; Burgess, M. A.; Plager, C.; Papadopolous, N.; Jenkins, J.; and Benjamin, R. S.: Results of two consecutive trials of dose-intensive chemotherapy with doxorubicin and ifosfamide in patients with sarcomas. Am J Clin Oncol, 21(3): 317-21, 1998.
37. Pisters, P. W.; Harrison, L. B.; Leung, D. H.; Woodruff, J. M.; Casper, E. S.; and Brennan, M. F.: Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol, 14(3): 859-68, 1996.
38. Pisters, P. W.; Harrison, L. B.; Woodruff, J. M.; Gaynor, J. J.; and Brennan, M. F.: A prospective randomized trial of adjuvant brachytherapy in the management of low-grade soft tissue sarcomas of the extremity and superficial trunk. J Clin Oncol, 12(6): 1150-5, 1994.
39. Pisters, P. W.; Leung, D. H.; Woodruff, J.; Shi, W.; and Brennan, M. F.: Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol, 14(5): 1679-89, 1996.
40. Rosenberg, S. A. et al.: The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg, 196(3): 305-15, 1982.
41. Roth, J. A.; Putnam, J. B., Jr.; Wesley, M. N.; and Rosenberg, S. A.: Differing determinants of prognosis following resection of pulmonary metastases from osteogenic and soft tissue sarcoma patients. Cancer, 55(6): 1361-6, 1985.
42. Salo, J. C.; Lewis, J. J.; Woodruff, J. M.; Leung, D. H.; and Brennan, M. F.: Malignant fibrous histiocytoma of the extremity. Cancer, 85(8): 1765-72, 1999.
43. Sindelar, W. F.; Kinsella, T. J.; Chen, P. W.; DeLaney, T. F.; Tepper, J. E.; Rosenberg, S. A.; and Glatstein, E.: Intraoperative radiotherapy in retroperitoneal sarcomas. Final results of a prospective, randomized, clinical trial. Arch Surg, 128(4): 402-10, 1993.
44. Singer, S.; Antman, K.; Corson, J. M.; and Eberlein, T. J.: Long-term salvageability for patients with locally recurrent soft-tissue sarcomas. Arch Surg, 127(5): 548-53; discussion 553-4, 1992.
45. Skrzynski, M. C.; Biermann, J. S.; Montag, A.; and Simon, M. A.: Diagnostic accuracy and charge-savings of outpatient core needle biopsy compared with open biopsy of musculoskeletal tumors. J Bone Joint Surg Am, 78(5): 644-9, 1996.
46. Williard, W. C.; Collin, C.; Casper, E. S.; Hajdu, S. I.; and Brennan, M. F.: The changing role of amputation for soft tissue sarcoma of the extremity in adults. Surg Gynecol Obstet, 175(5): 389-96, 1992.
47. Williard, W. C.; Hajdu, S. I.; Casper, E. S.; and Brennan, M. F.: Comparison of amputation with limb-sparing operations for adult soft tissue sarcoma of the extremity. Ann Surg, 215(3): 269-75, 1992.
48. Yang, J. C. et al.: Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol, 16(1): 197-203, 1998.
49. Zagars, G. K.; Mullen, J. R.; and Pollack, A.: Malignant fibrous histiocytoma: outcome and prognostic factors following conservation surgery and radiotherapy. Int J Radiat Oncol Biol Phys, 34(5): 983-94, 1996.
